

Within-Host Evolution of Human Influenza Virus
- PMID: 29534854
- PMCID: PMC6097882
- DOI: 10.1016/j.tim.2018.02.007
Within-Host Evolution of Human Influenza Virus
Abstract
The rapid global evolution of influenza virus begins with mutations that arise de novo in individual infections, but little is known about how evolution occurs within hosts. We review recent progress in understanding how and why influenza viruses evolve within human hosts. Advances in deep sequencing make it possible to measure within-host genetic diversity in both acute and chronic influenza infections. Factors like antigenic selection, antiviral treatment, tissue specificity, spatial structure, and multiplicity of infection may affect how influenza viruses evolve within human hosts. Studies of within-host evolution can contribute to our understanding of the evolutionary and epidemiological factors that shape influenza virus's global evolution.
Keywords: deep sequencing; evolution; influenza; within-host.
Copyright © 2018 Elsevier Ltd. All rights reserved.
Figures
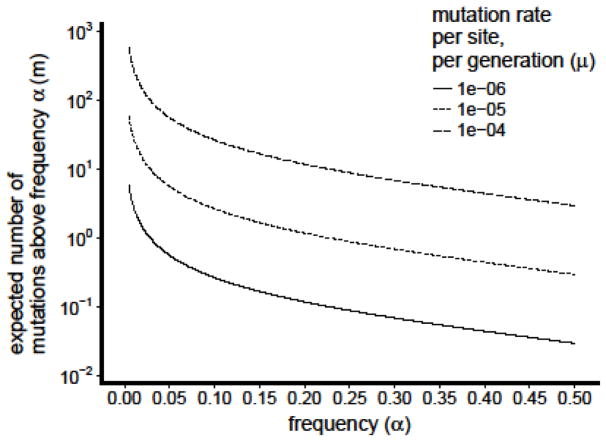
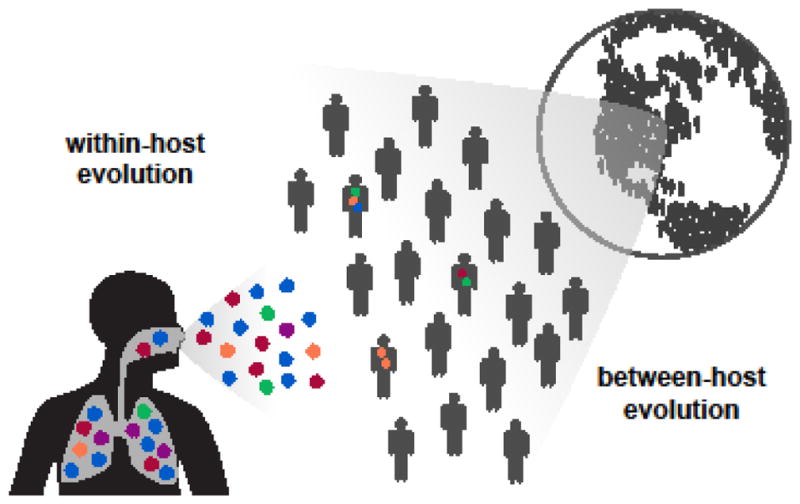
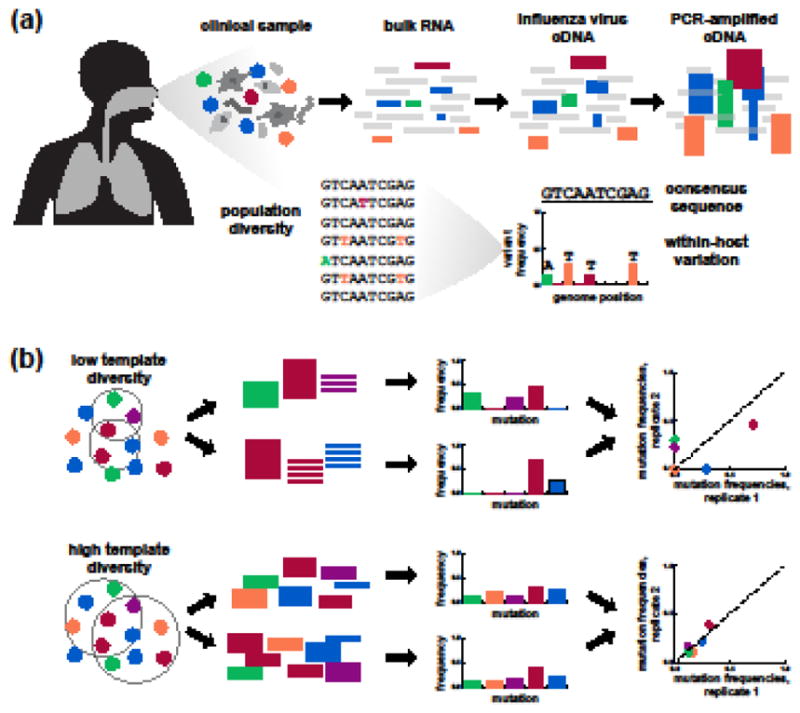
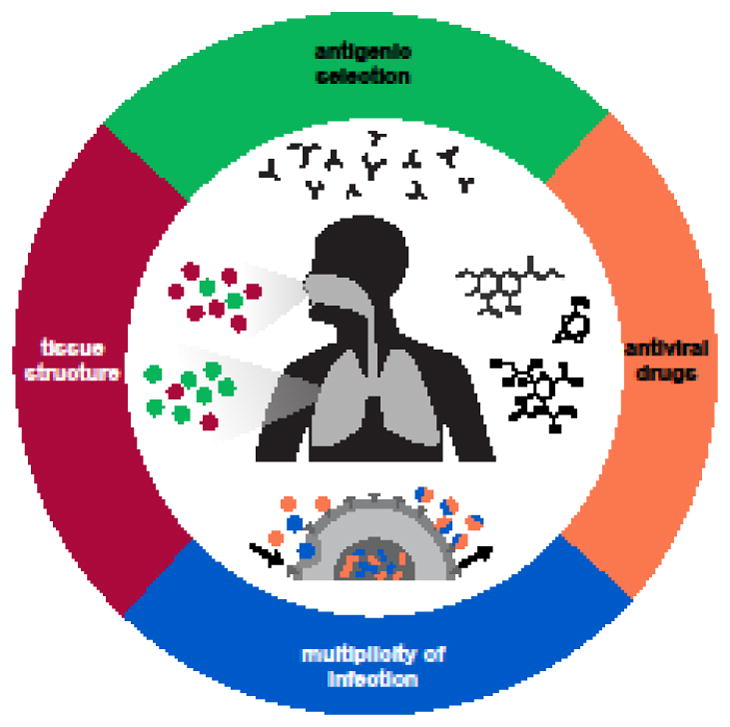

References
-
- Ghedin E, et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature. 2005;437:1162–1166. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
