

MetaGO: Predicting Gene Ontology of Non-homologous Proteins Through Low-Resolution Protein Structure Prediction and Protein-Protein Network Mapping
- PMID: 29534977
- PMCID: PMC6014880
- DOI: 10.1016/j.jmb.2018.03.004
MetaGO: Predicting Gene Ontology of Non-homologous Proteins Through Low-Resolution Protein Structure Prediction and Protein-Protein Network Mapping
Abstract
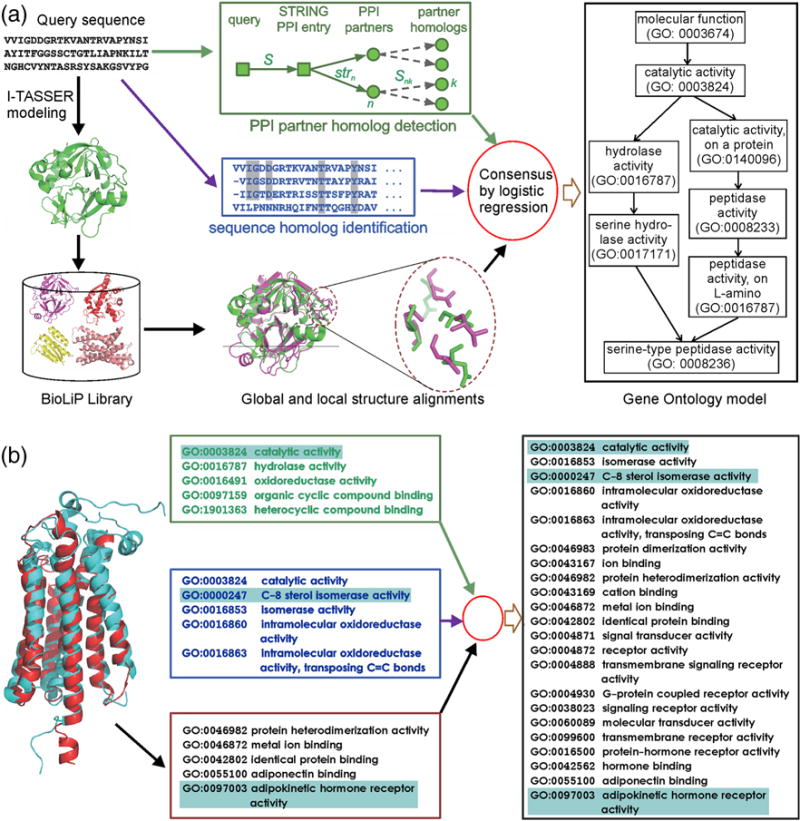
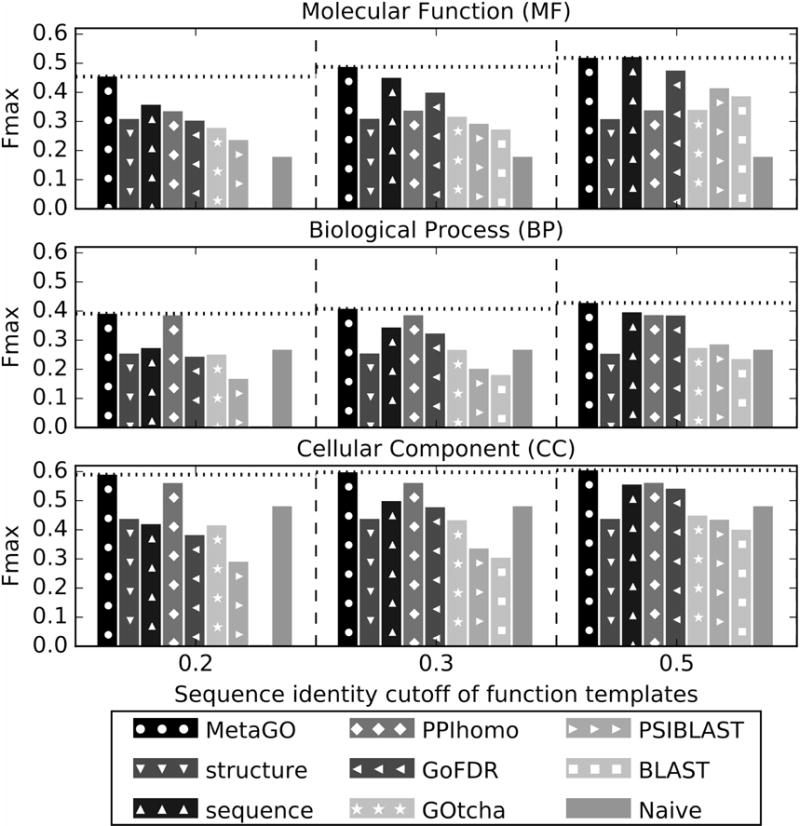
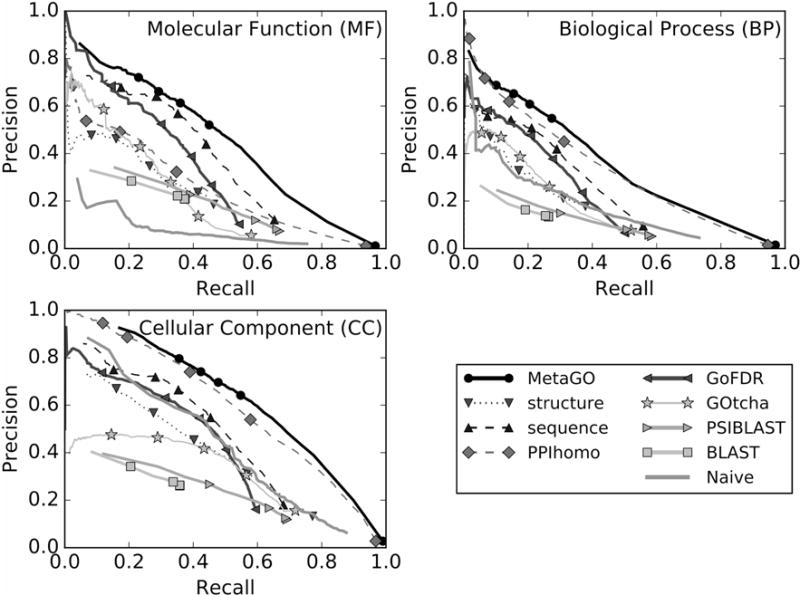
Homology-based transferal remains the major approach to computational protein function annotations, but it becomes increasingly unreliable when the sequence identity between query and template decreases below 30%. We propose a novel pipeline, MetaGO, to deduce Gene Ontology attributes of proteins by combining sequence homology-based annotation with low-resolution structure prediction and comparison, and partner's homology-based protein-protein network mapping. The pipeline was tested on a large-scale set of 1000 non-redundant proteins from the CAFA3 experiment. Under the stringent benchmark conditions where templates with >30% sequence identity to the query are excluded, MetaGO achieves average F-measures of 0.487, 0.408, and 0.598, for Molecular Function, Biological Process, and Cellular Component, respectively, which are significantly higher than those achieved by other state-of-the-art function annotations methods. Detailed data analysis shows that the major advantage of the MetaGO lies in the new functional homolog detections from partner's homology-based network mapping and structure-based local and global structure alignments, the confidence scores of which can be optimally combined through logistic regression. These data demonstrate the power of using a hybrid model incorporating protein structure and interaction networks to deduce new functional insights beyond traditional sequence homology-based referrals, especially for proteins that lack homologous function templates. The MetaGO pipeline is available at http://zhanglab.ccmb.med.umich.edu/MetaGO/.
Keywords: Gene Ontology; protein function prediction; protein structure prediction; protein–protein interaction; sequence profiles.
Copyright © 2018 Elsevier Ltd. All rights reserved.
Figures
References
-
- Hirschhorn JN. Genomewide Association Studies - Illuminating Biologic Pathways. New Engl J Med. 2009;360:1699–701. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
