

Defining the Transcriptional Landscape during Cytomegalovirus Latency with Single-Cell RNA Sequencing
- PMID: 29535194
- PMCID: PMC5850328
- DOI: 10.1128/mBio.00013-18
Defining the Transcriptional Landscape during Cytomegalovirus Latency with Single-Cell RNA Sequencing
Abstract
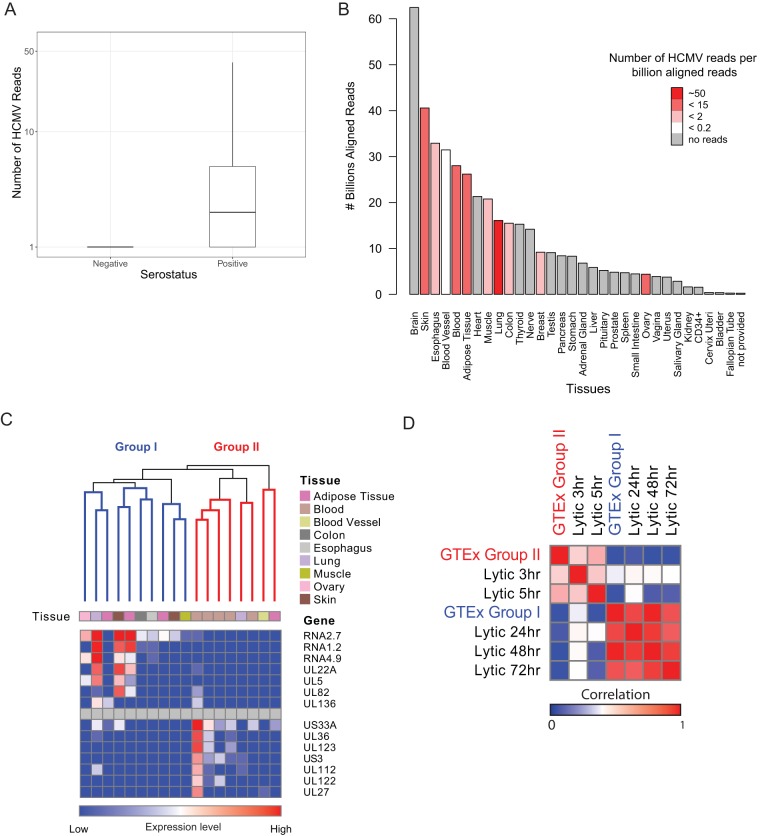
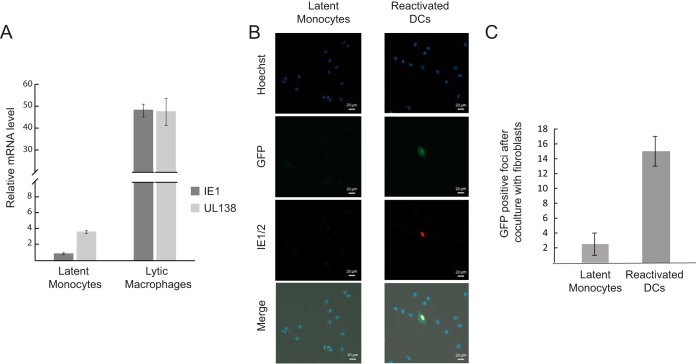
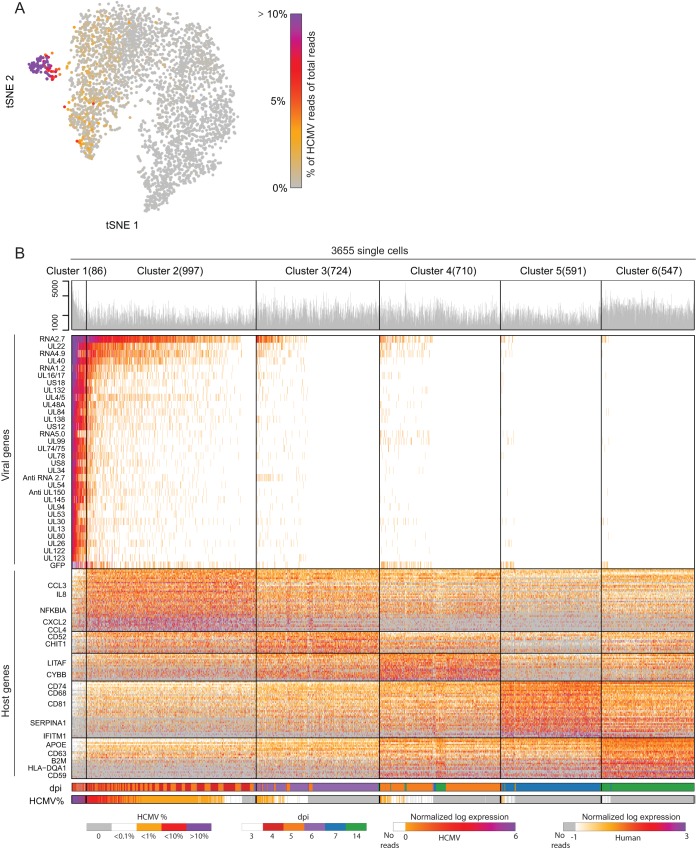
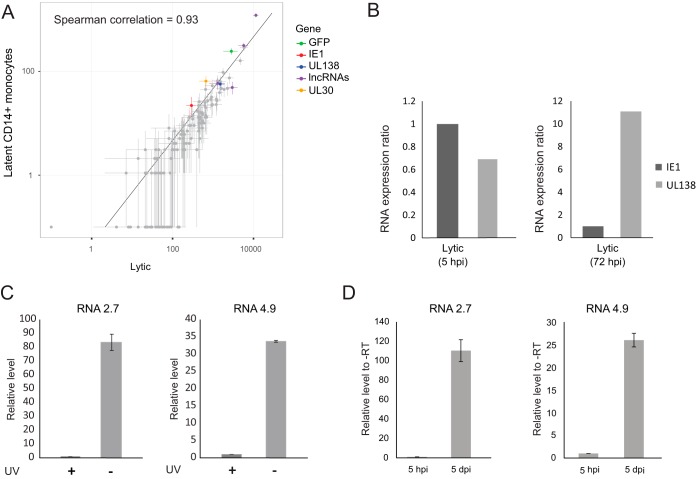
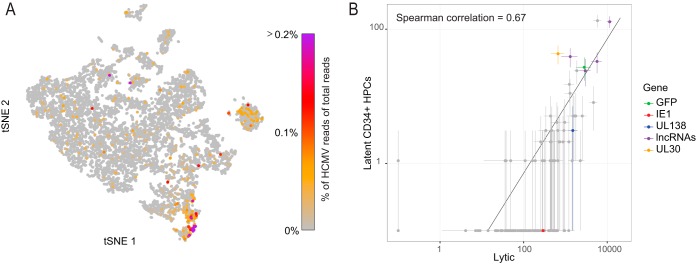
Primary infection with human cytomegalovirus (HCMV) results in a lifelong infection due to its ability to establish latent infection, with one characterized viral reservoir being hematopoietic cells. Although reactivation from latency causes serious disease in immunocompromised individuals, our molecular understanding of latency is limited. Here, we delineate viral gene expression during natural HCMV persistent infection by analyzing the massive transcriptome RNA sequencing (RNA-seq) atlas generated by the Genotype-Tissue Expression (GTEx) project. This systematic analysis reveals that HCMV persistence in vivo is prevalent in diverse tissues. Notably, we find only viral transcripts that resemble gene expression during various stages of lytic infection with no evidence of any highly restricted latency-associated viral gene expression program. To further define the transcriptional landscape during HCMV latent infection, we also used single-cell RNA-seq and a tractable experimental latency model. In contrast to some current views on latency, we also find no evidence for any highly restricted latency-associated viral gene expression program. Instead, we reveal that latency-associated gene expression largely mirrors a late lytic viral program, albeit at much lower levels of expression. Overall, our work has the potential to revolutionize our understanding of HCMV persistence and suggests that latency is governed mainly by quantitative changes, with a limited number of qualitative changes, in viral gene expression.IMPORTANCE Human cytomegalovirus is a prevalent pathogen, infecting most of the population worldwide and establishing lifelong latency in its hosts. Although reactivation from latency causes significant morbidity and mortality in immunocompromised hosts, our molecular understanding of the latent state remains limited. Here, we examine the viral gene expression during natural and experimental latent HCMV infection on a transcriptome-wide level. In contrast to the classical views on herpesvirus latency, we find no evidence for a restricted latency-associated viral gene expression program. Instead, we reveal that latency gene expression largely resembles a late lytic viral profile, albeit at much lower levels of expression. Taken together, our data transform the current view of HCMV persistence and suggest that latency is mainly governed by quantitative rather than qualitative changes in viral gene expression.
Keywords: cytomegalovirus; gene expression; latency; single-cell RNA-seq; transcriptome.
Copyright © 2018 Shnayder et al.
Figures
Comment in
-
A Single-Cell Approach to the Elusive Latent Human Cytomegalovirus Transcriptome.mBio. 2018 Jun 12;9(3):e01001-18. doi: 10.1128/mBio.01001-18. mBio. 2018. PMID: 29895640 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
