

Benchmarking common quantification strategies for large-scale phosphoproteomics
- PMID: 29535314
- PMCID: PMC5849679
- DOI: 10.1038/s41467-018-03309-6
Benchmarking common quantification strategies for large-scale phosphoproteomics
Abstract
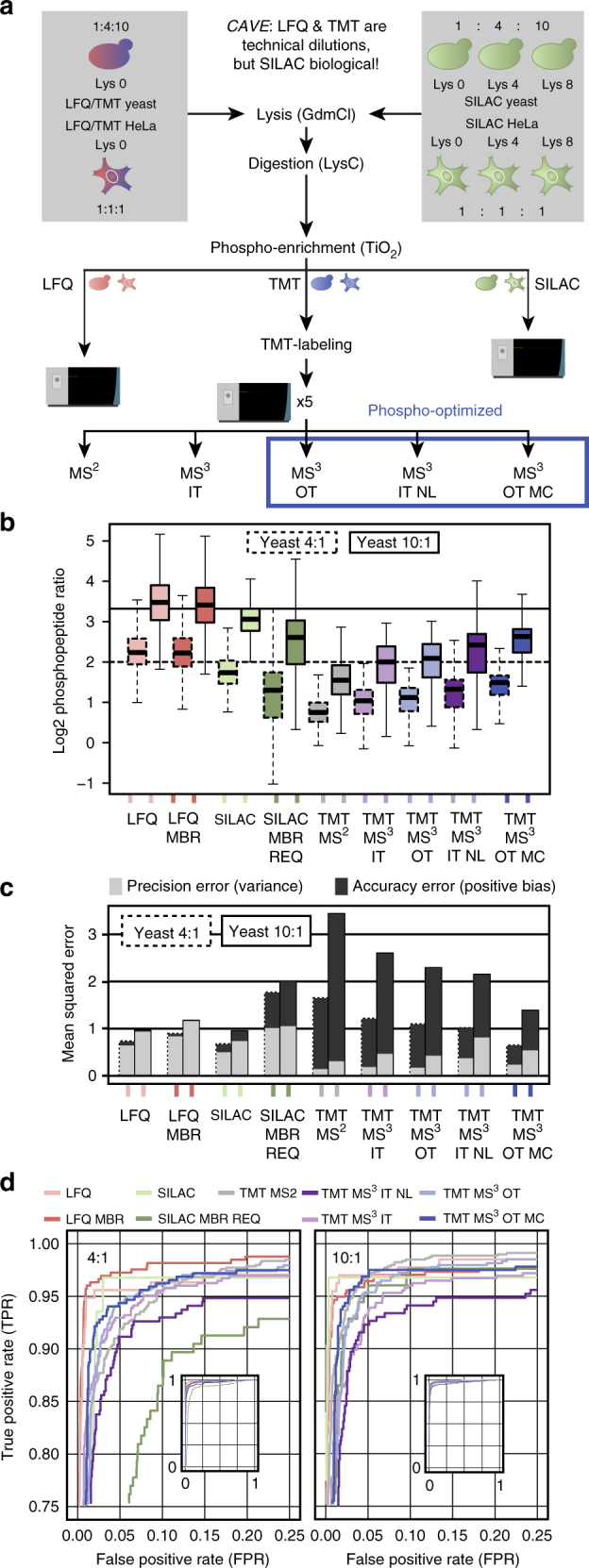
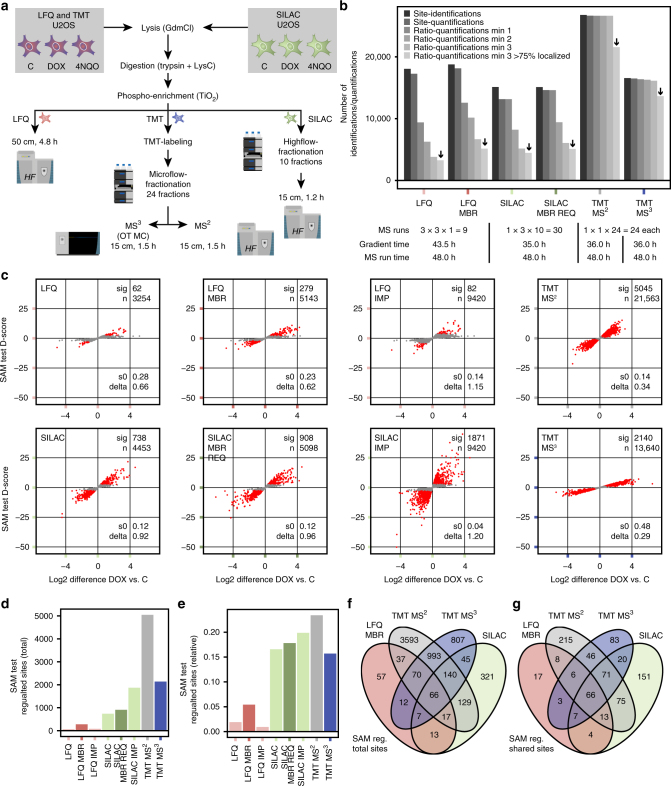
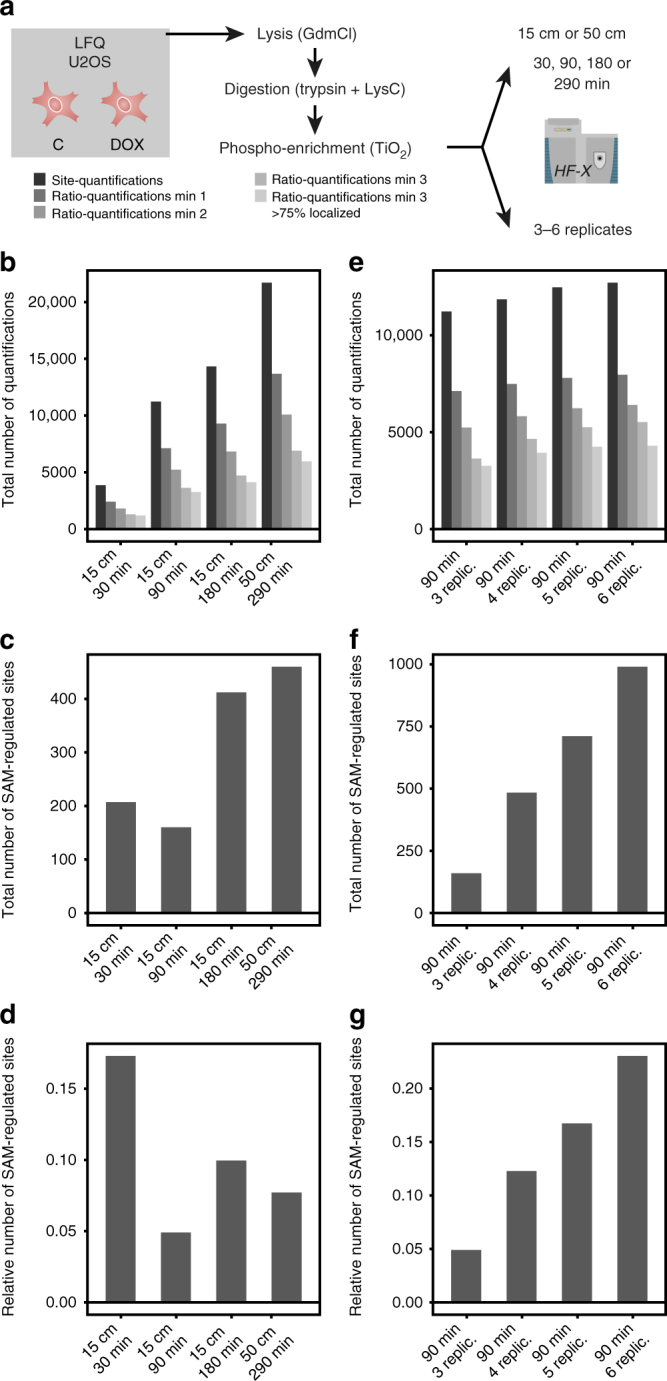
Comprehensive mass spectrometry (MS)-based proteomics is now feasible, but reproducible quantification remains challenging, especially for post-translational modifications such as phosphorylation. Here, we compare the most popular quantification techniques for global phosphoproteomics: label-free quantification (LFQ), stable isotope labeling by amino acids in cell culture (SILAC) and MS2- and MS3-measured tandem mass tags (TMT). In a mixed species comparison with fixed phosphopeptide ratios, we find LFQ and SILAC to be the most accurate techniques. MS2-based TMT yields the highest precision but lowest accuracy due to ratio compression, which MS3-based TMT can partly rescue. However, MS2-based TMT outperforms MS3-based TMT when analyzing phosphoproteome changes in the DNA damage response, since its higher precision and larger identification numbers allow detection of a greater number of significantly regulated phosphopeptides. Finally, we utilize the TMT multiplexing capabilities to develop an algorithm for determining phosphorylation site stoichiometry, showing that such applications benefit from the high accuracy of MS3-based TMT.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
