

Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy
- PMID: 29538625
- PMCID: PMC5917748
- DOI: 10.1093/brain/awy046
Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy
Abstract
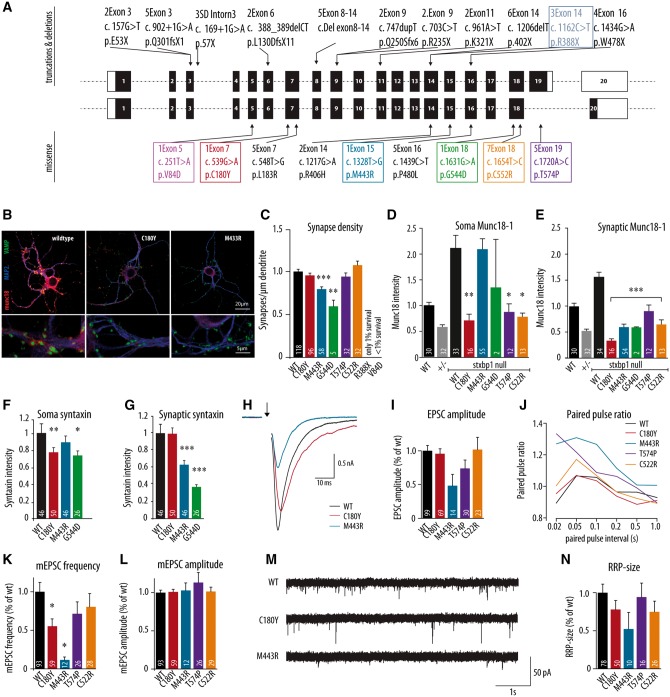
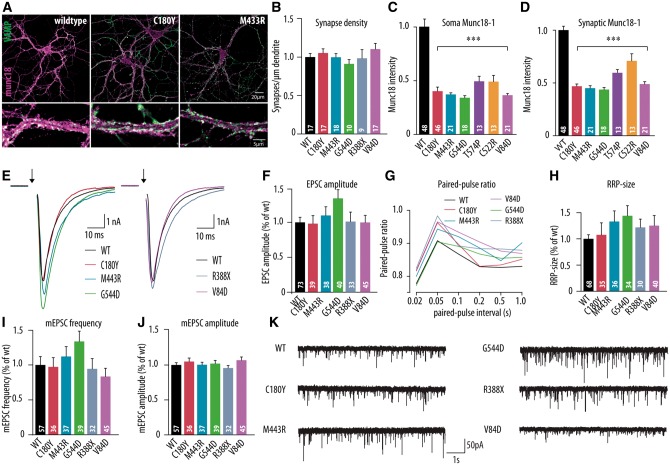
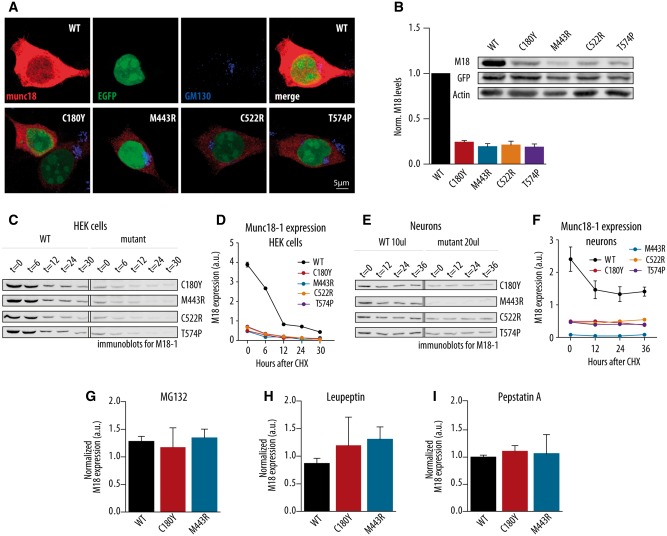
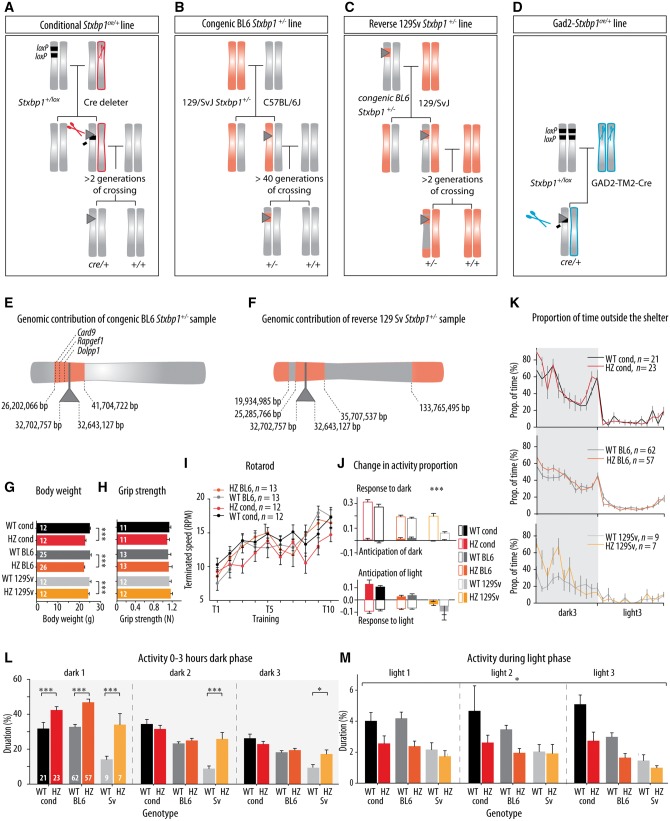
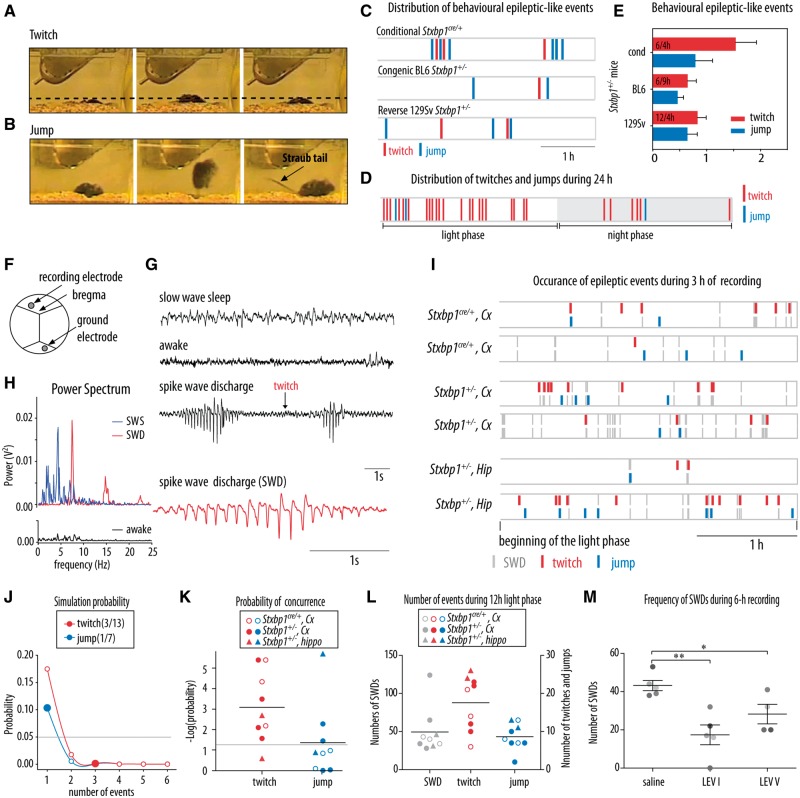
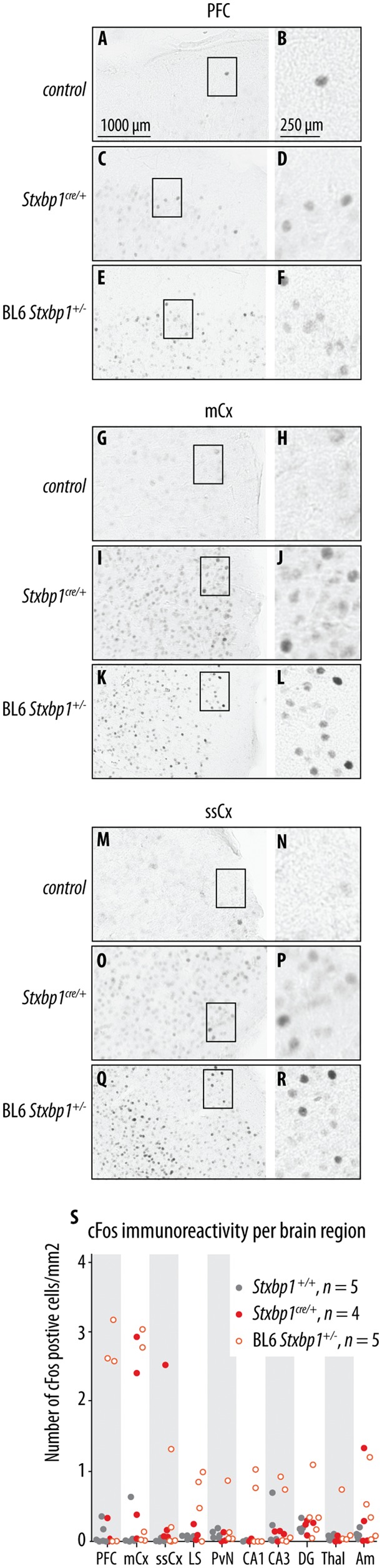
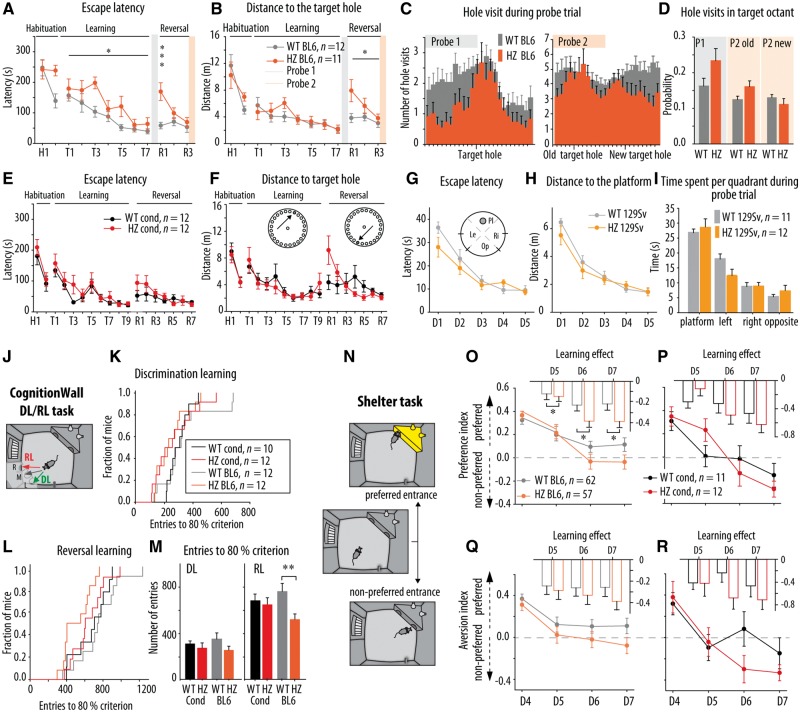
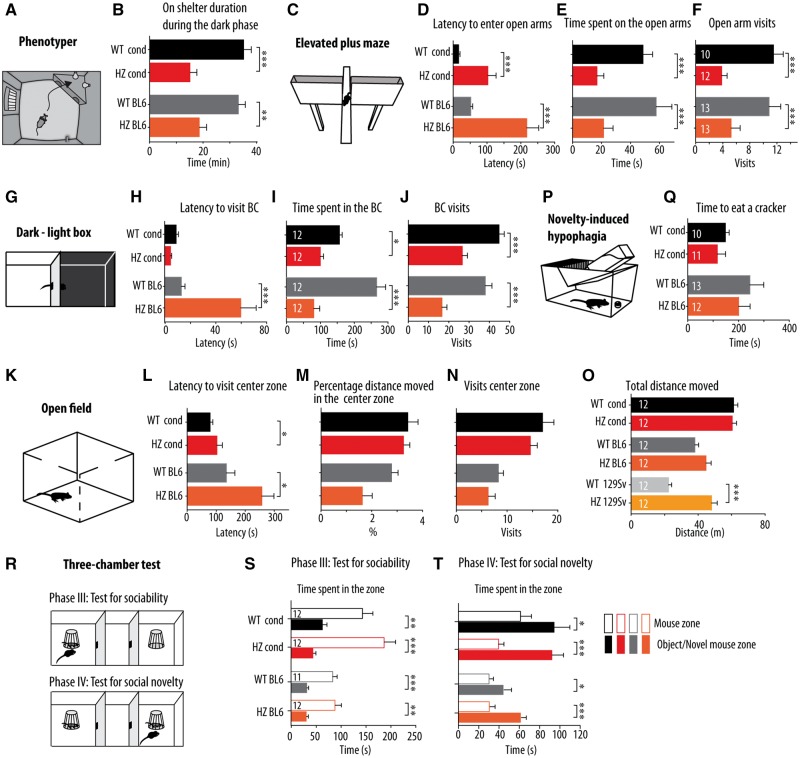
De novo heterozygous mutations in STXBP1/Munc18-1 cause early infantile epileptic encephalopathies (EIEE4, OMIM #612164) characterized by infantile epilepsy, developmental delay, intellectual disability, and can include autistic features. We characterized the cellular deficits for an allelic series of seven STXBP1 mutations and developed four mouse models that recapitulate the abnormal EEG activity and cognitive aspects of human STXBP1-encephalopathy. Disease-causing STXBP1 variants supported synaptic transmission to a variable extent on a null background, but had no effect when overexpressed on a heterozygous background. All disease variants had severely decreased protein levels. Together, these cellular studies suggest that impaired protein stability and STXBP1 haploinsufficiency explain STXBP1-encephalopathy and that, therefore, Stxbp1+/- mice provide a valid mouse model. Simultaneous video and EEG recordings revealed that Stxbp1+/- mice with different genomic backgrounds recapitulate the seizure/spasm phenotype observed in humans, characterized by myoclonic jerks and spike-wave discharges that were suppressed by the antiepileptic drug levetiracetam. Mice heterozygous for Stxbp1 in GABAergic neurons only, showed impaired viability, 50% died within 2-3 weeks, and the rest showed stronger epileptic activity. c-Fos staining implicated neocortical areas, but not other brain regions, as the seizure foci. Stxbp1+/- mice showed impaired cognitive performance, hyperactivity and anxiety-like behaviour, without altered social behaviour. Taken together, these data demonstrate the construct, face and predictive validity of Stxbp1+/- mice and point to protein instability, haploinsufficiency and imbalanced excitation in neocortex, as the underlying mechanism of STXBP1-encephalopathy. The mouse models reported here are valid models for development of therapeutic interventions targeting STXBP1-encephalopathy.
Figures
Comment in
-
Trading up to a New Model of STXBP-Encephalopathy.Epilepsy Curr. 2018 Jul-Aug;18(4):257-259. doi: 10.5698/1535-7597.18.4.257. Epilepsy Curr. 2018. PMID: 30254526 Free PMC article. No abstract available.
References
-
- Barcia G, Chemaly N, Gobin S, Milh M, Van Bogaert P, Barnerias C, et al.Early epileptic encephalopathies associated with STXBP1 mutations: could we better delineate the phenotype? Eur J Med Genet 2014; 57: 15–20. - PubMed
-
- Beyenburg S, Mitchell AJ, Schmidt D, Elger CE, Reuber M. Anxiety in patients with epilepsy: systematic review and suggestions for clinical management [Review]. Epilepsy Behav 2005; 7: 161–71. - PubMed
-
- Brose N, O'Connor V, Skehel P. Synaptopathy: dysfunction of synaptic function? Biochem Soc Trans 2010; 38: 443–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
