

DROIDS 1.20: A GUI-Based Pipeline for GPU-Accelerated Comparative Protein Dynamics
- PMID: 29539389
- PMCID: PMC5883555
- DOI: 10.1016/j.bpj.2018.01.020
DROIDS 1.20: A GUI-Based Pipeline for GPU-Accelerated Comparative Protein Dynamics
Abstract
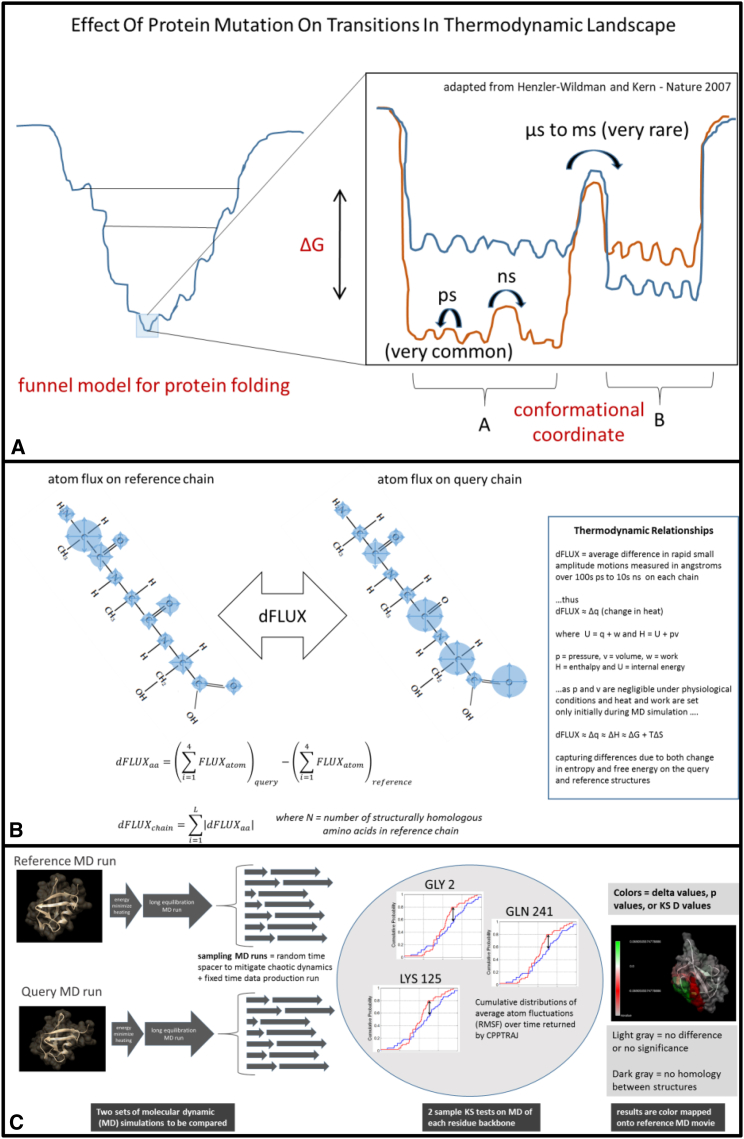
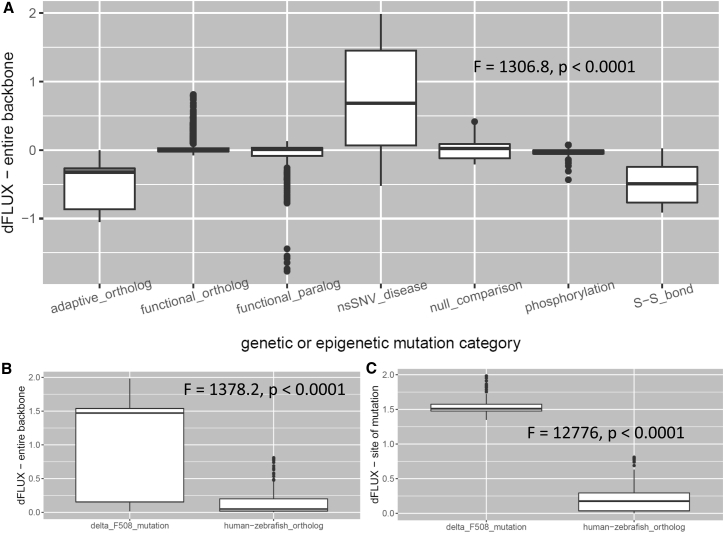
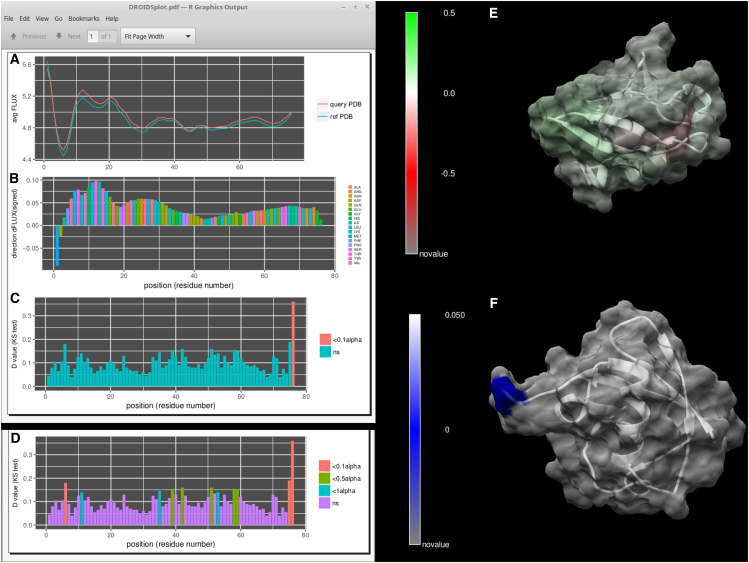
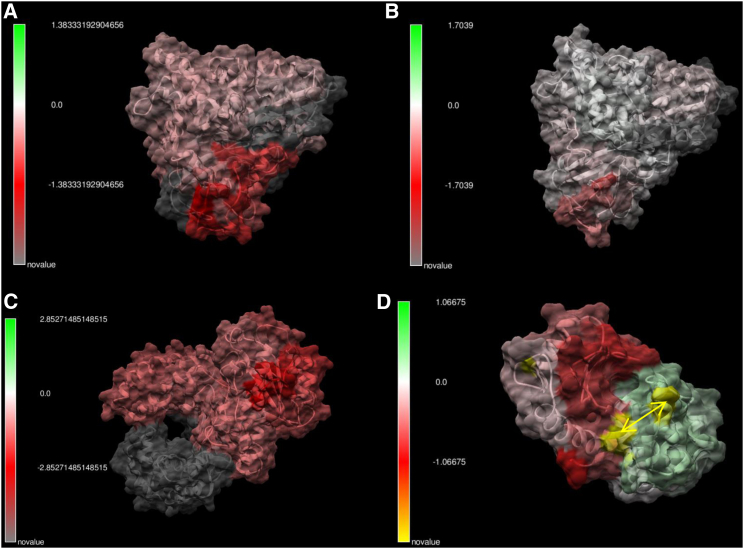
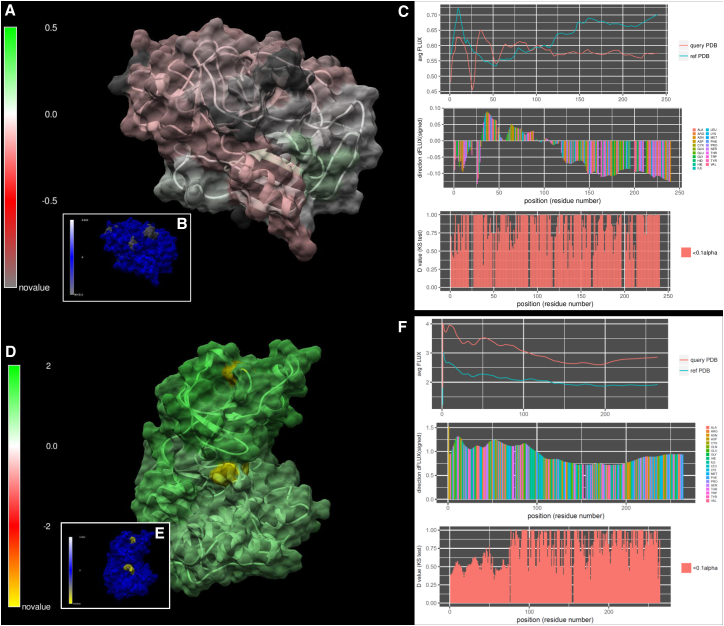
Traditional informatics in comparative genomics work only with static representations of biomolecules (i.e., sequence and structure), thereby ignoring the molecular dynamics (MD) of proteins that define function in the cell. A comparative approach applied to MD would connect this very short timescale process, defined in femtoseconds, to one of the longest in the universe: molecular evolution measured in millions of years. Here, we leverage advances in graphics-processing-unit-accelerated MD simulation software to develop a comparative method of MD analysis and visualization that can be applied to any two homologous Protein Data Bank structures. Our open-source pipeline, DROIDS (Detecting Relative Outlier Impacts in Dynamic Simulations), works in conjunction with existing molecular modeling software to convert any Linux gaming personal computer into a "comparative computational microscope" for observing the biophysical effects of mutations and other chemical changes in proteins. DROIDS implements structural alignment and Benjamini-Hochberg-corrected Kolmogorov-Smirnov statistics to compare nanosecond-scale atom bond fluctuations on the protein backbone, color mapping the significant differences identified in protein MD with single-amino-acid resolution. DROIDS is simple to use, incorporating graphical user interface control for Amber16 MD simulations, cpptraj analysis, and the final statistical and visual representations in R graphics and UCSF Chimera. We demonstrate that DROIDS can be utilized to visually investigate molecular evolution and disease-related functional changes in MD due to genetic mutation and epigenetic modification. DROIDS can also be used to potentially investigate binding interactions of pharmaceuticals, toxins, or other biomolecules in a functional evolutionary context as well.
Copyright © 2018 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Babbitt G.A., Coppola E.E., Hudson A.O. Can all heritable biology really be reduced to a single dimension? Gene. 2016;578:162–168. - PubMed
-
- Salomon-Ferrer R., Götz A.W., Walker R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013;9:3878–3888. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
