

The cis-regulatory dynamics of embryonic development at single-cell resolution
- PMID: 29539636
- PMCID: PMC5866720
- DOI: 10.1038/nature25981
The cis-regulatory dynamics of embryonic development at single-cell resolution
Abstract
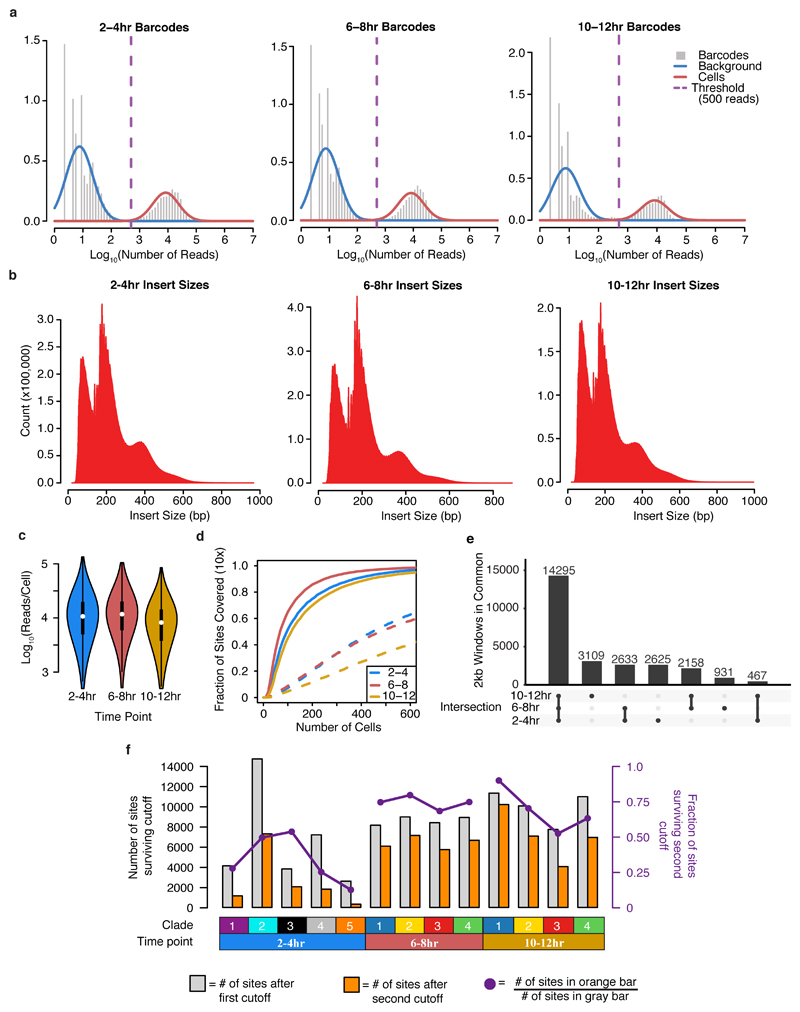
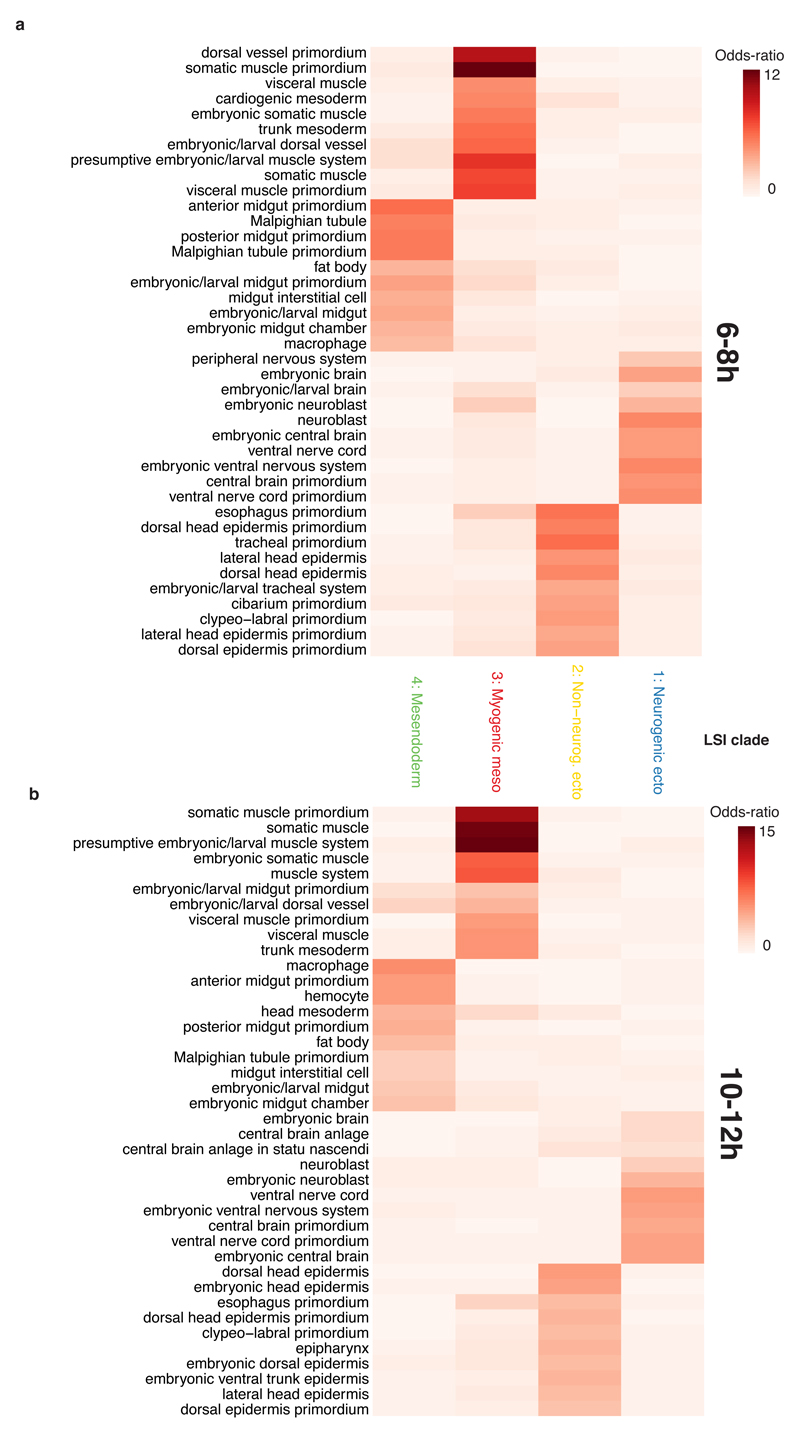
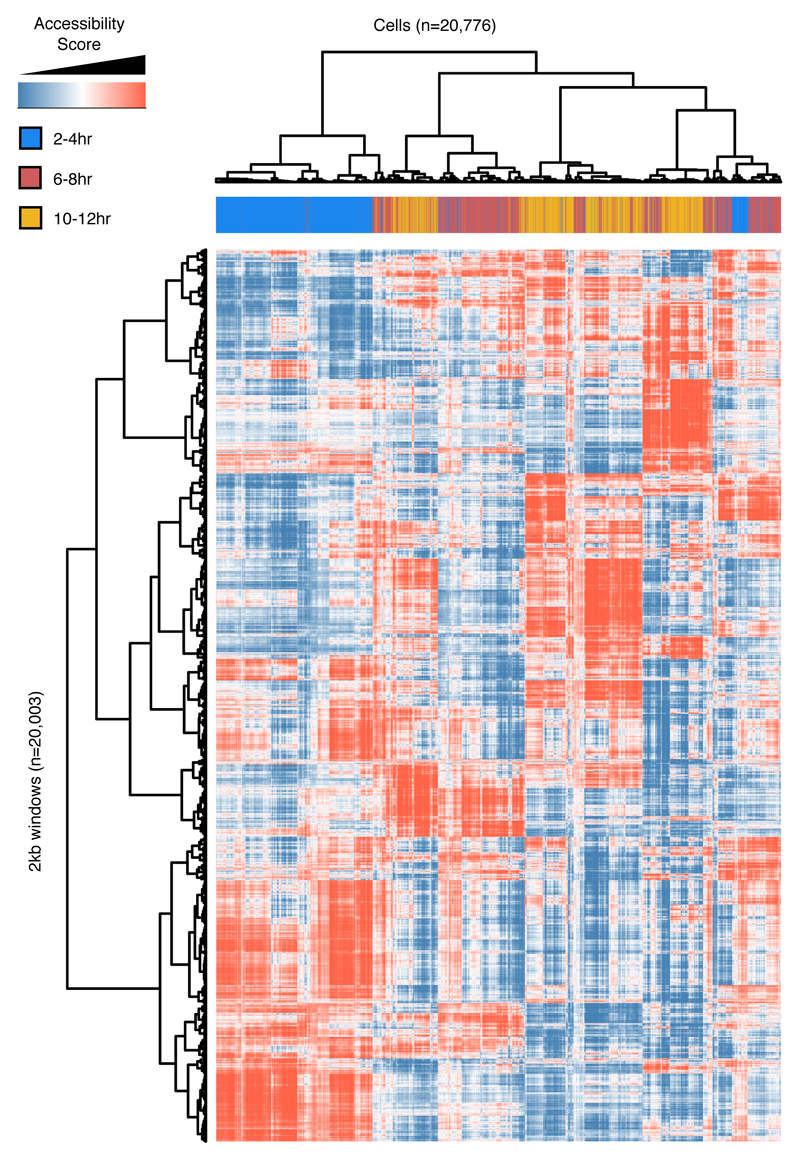
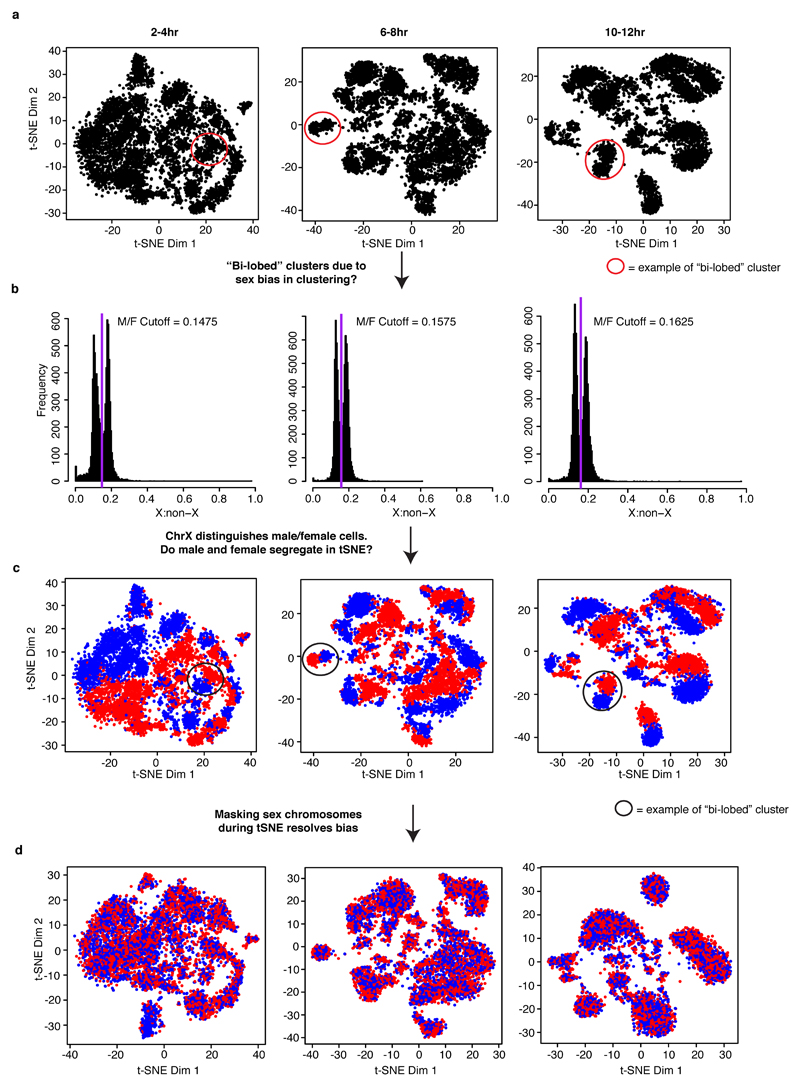
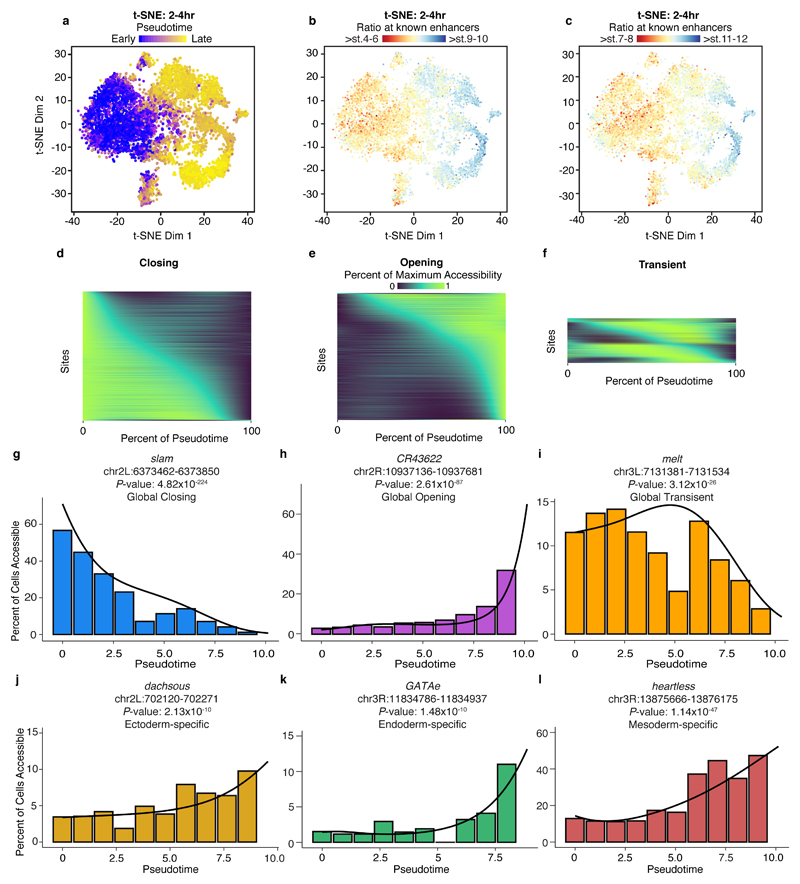
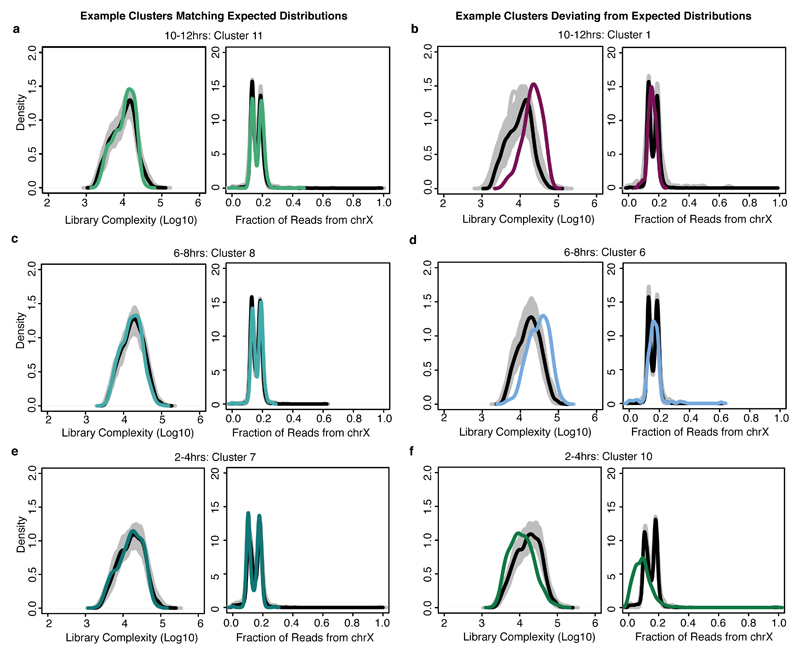
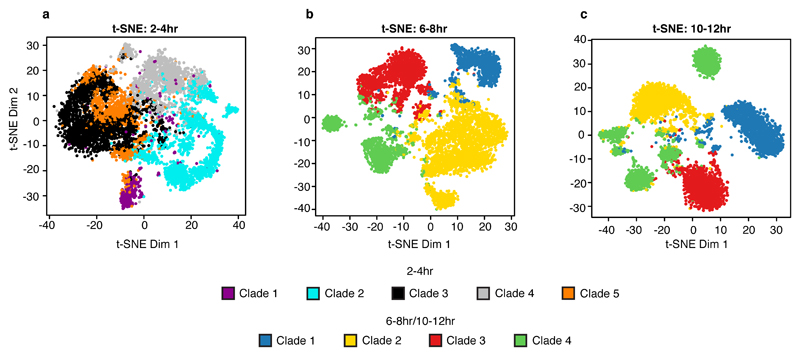
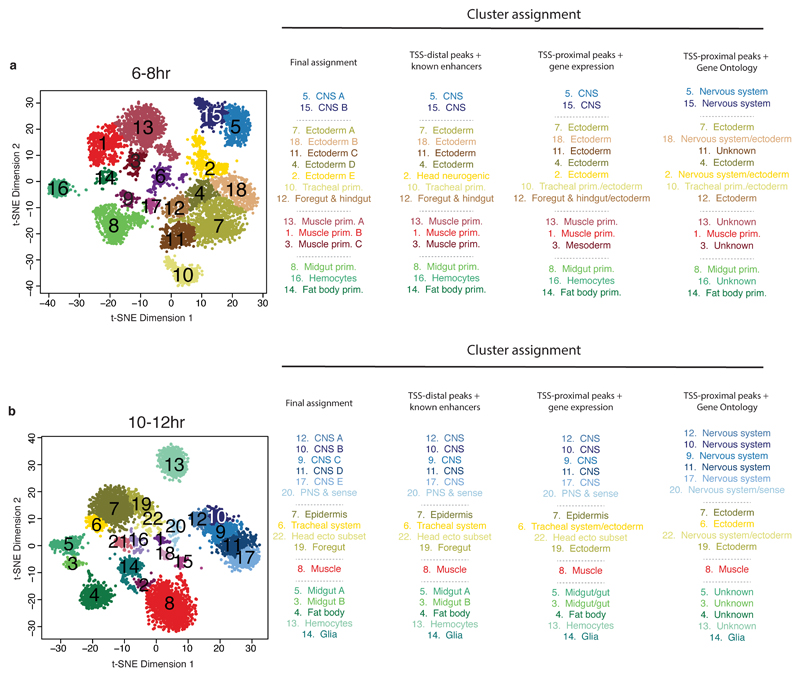
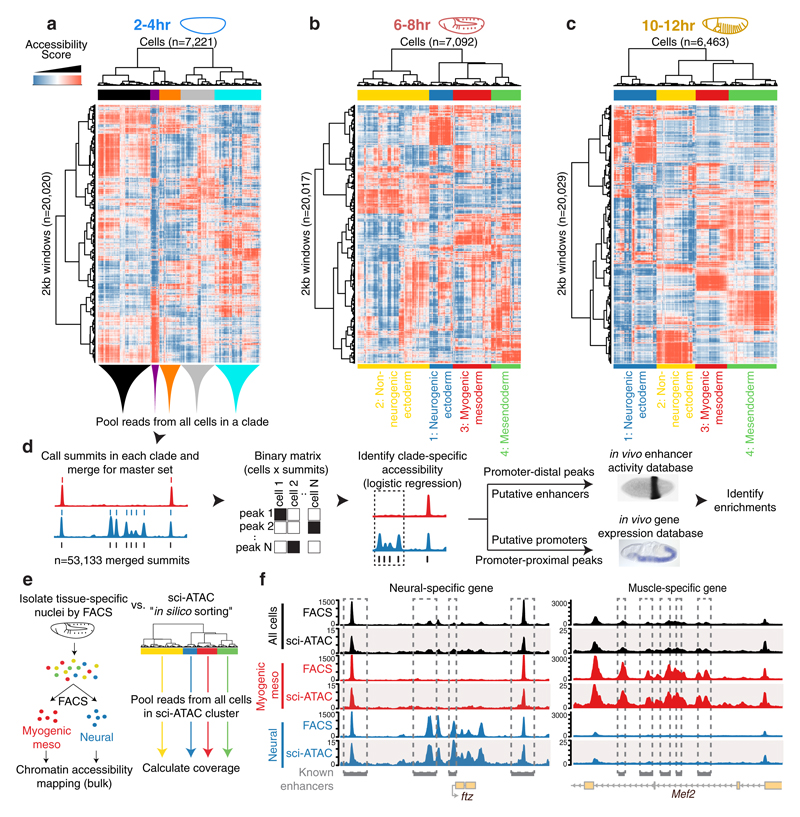
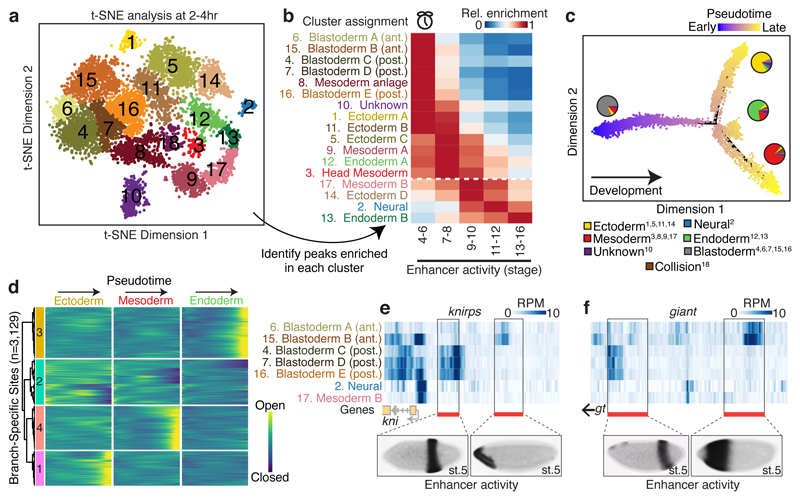
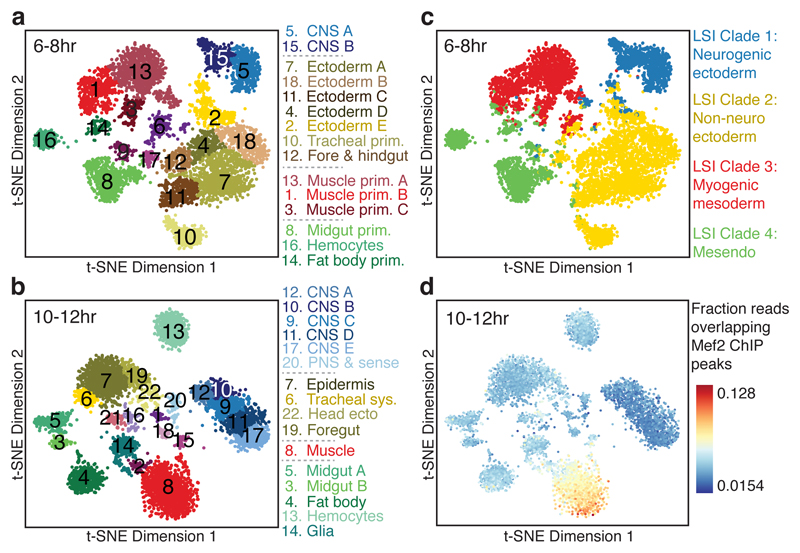
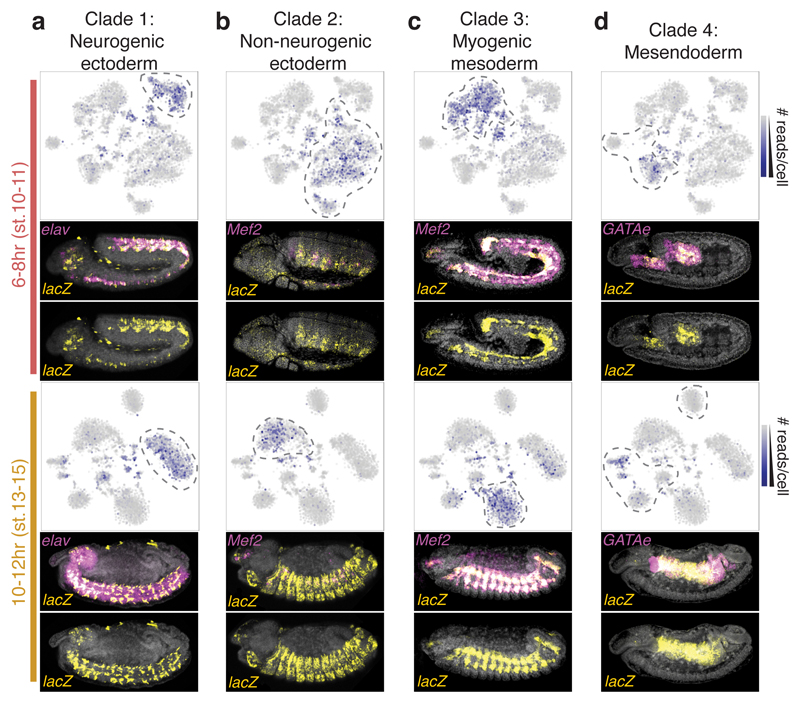
Understanding how gene regulatory networks control the progressive restriction of cell fates is a long-standing challenge. Recent advances in measuring gene expression in single cells are providing new insights into lineage commitment. However, the regulatory events underlying these changes remain unclear. Here we investigate the dynamics of chromatin regulatory landscapes during embryogenesis at single-cell resolution. Using single-cell combinatorial indexing assay for transposase accessible chromatin with sequencing (sci-ATAC-seq), we profiled chromatin accessibility in over 20,000 single nuclei from fixed Drosophila melanogaster embryos spanning three landmark embryonic stages: 2-4 h after egg laying (predominantly stage 5 blastoderm nuclei), when each embryo comprises around 6,000 multipotent cells; 6-8 h after egg laying (predominantly stage 10-11), to capture a midpoint in embryonic development when major lineages in the mesoderm and ectoderm are specified; and 10-12 h after egg laying (predominantly stage 13), when each of the embryo's more than 20,000 cells are undergoing terminal differentiation. Our results show that there is spatial heterogeneity in the accessibility of the regulatory genome before gastrulation, a feature that aligns with future cell fate, and that nuclei can be temporally ordered along developmental trajectories. During mid-embryogenesis, tissue granularity emerges such that individual cell types can be inferred by their chromatin accessibility while maintaining a signature of their germ layer of origin. Analysis of the data reveals overlapping usage of regulatory elements between cells of the endoderm and non-myogenic mesoderm, suggesting a common developmental program that is reminiscent of the mesendoderm lineage in other species. We identify 30,075 distal regulatory elements that exhibit tissue-specific accessibility. We validated the germ-layer specificity of a subset of these predicted enhancers in transgenic embryos, achieving an accuracy of 90%. Overall, our results demonstrate the power of shotgun single-cell profiling of embryos to resolve dynamic changes in the chromatin landscape during development, and to uncover the cis-regulatory programs of metazoan germ layers and cell types.
Figures


References
-
- Maduro MF, Meneghini MD, Bowerman B, Broitman-Maduro G, Rothman JH. Restriction of mesendoderm to a single blastomere by the combined action of SKN-1 and a GSK-3beta homolog is mediated by MED-1 and -2 in C. elegans. Mol Cell. 2001;7:475–485. - PubMed
-
- Rodaway A, Patient R. Mesendoderm. an ancient germ layer? Cell. 2001;105:169–172. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
