

DNA methylation-based classification of central nervous system tumours
- PMID: 29539639
- PMCID: PMC6093218
- DOI: 10.1038/nature26000
DNA methylation-based classification of central nervous system tumours
Abstract
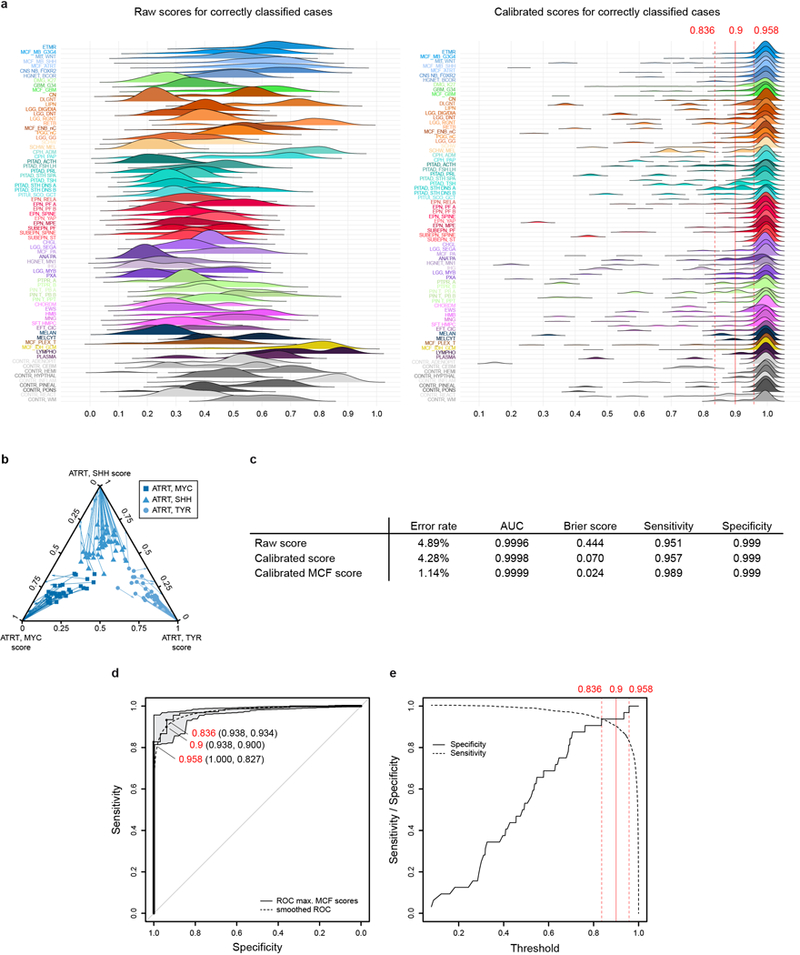
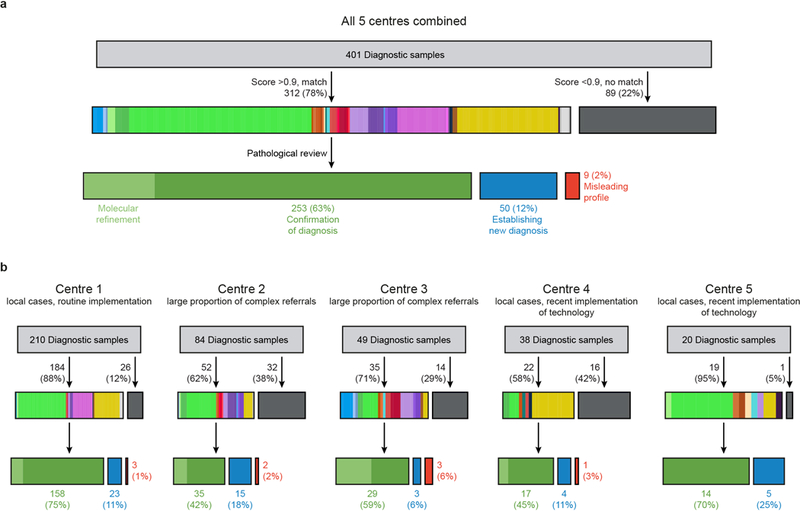
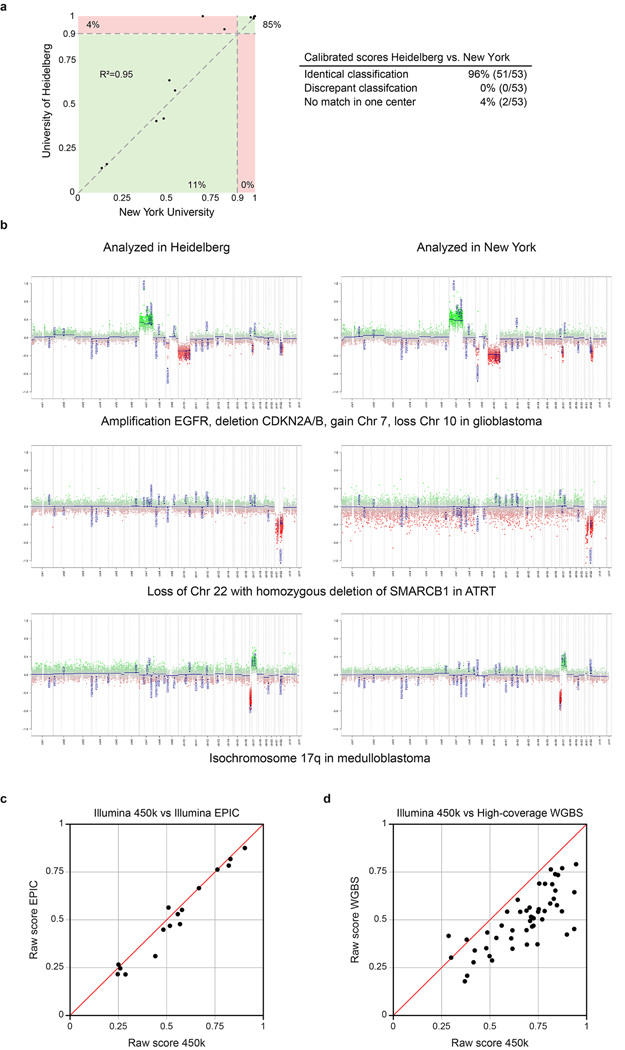
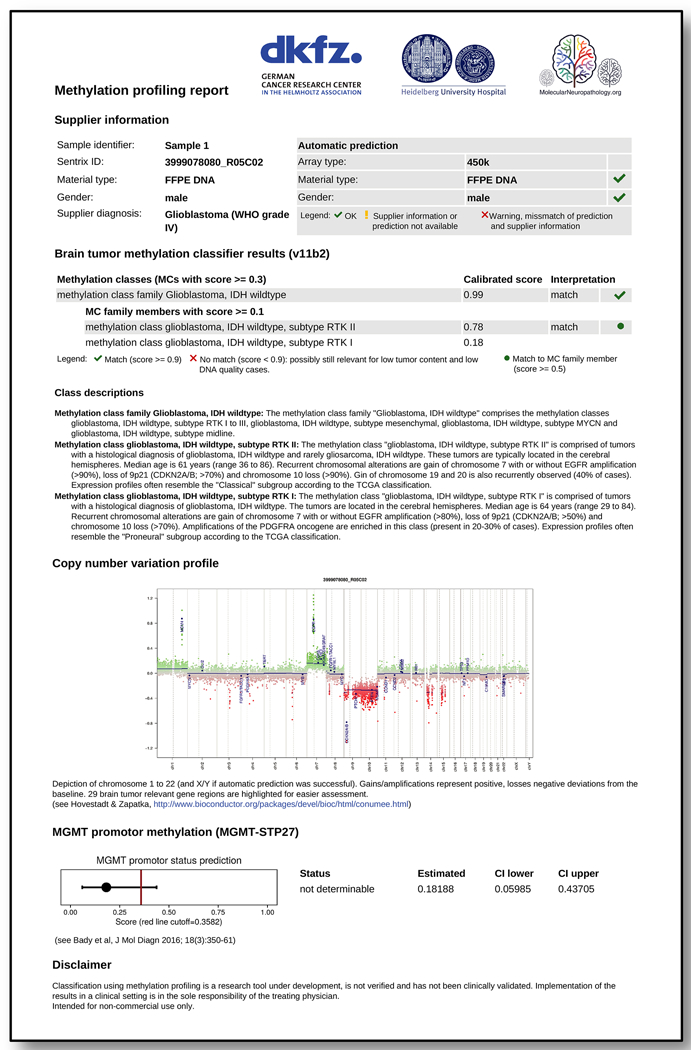
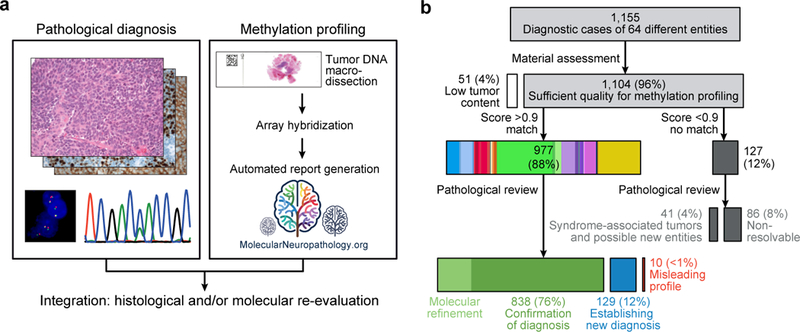
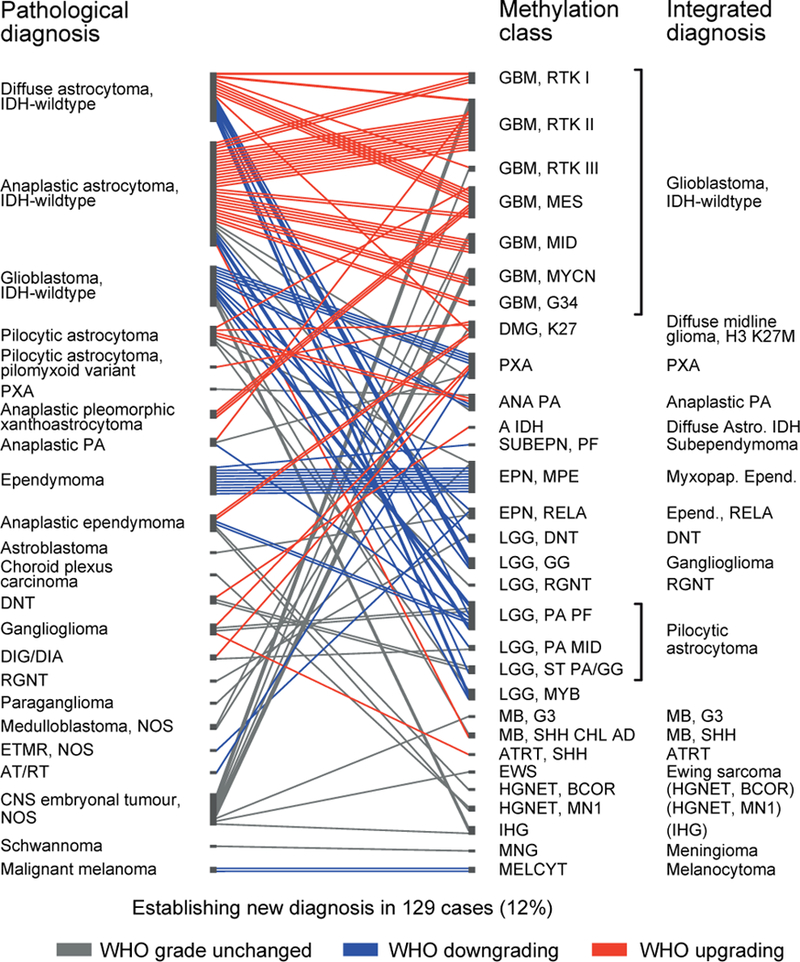
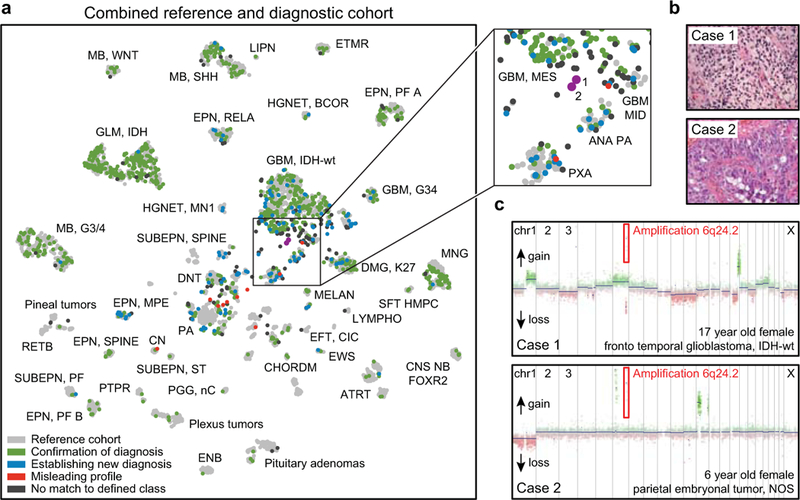
Accurate pathological diagnosis is crucial for optimal management of patients with cancer. For the approximately 100 known tumour types of the central nervous system, standardization of the diagnostic process has been shown to be particularly challenging-with substantial inter-observer variability in the histopathological diagnosis of many tumour types. Here we present a comprehensive approach for the DNA methylation-based classification of central nervous system tumours across all entities and age groups, and demonstrate its application in a routine diagnostic setting. We show that the availability of this method may have a substantial impact on diagnostic precision compared to standard methods, resulting in a change of diagnosis in up to 12% of prospective cases. For broader accessibility, we have designed a free online classifier tool, the use of which does not require any additional onsite data processing. Our results provide a blueprint for the generation of machine-learning-based tumour classifiers across other cancer entities, with the potential to fundamentally transform tumour pathology.
Figures
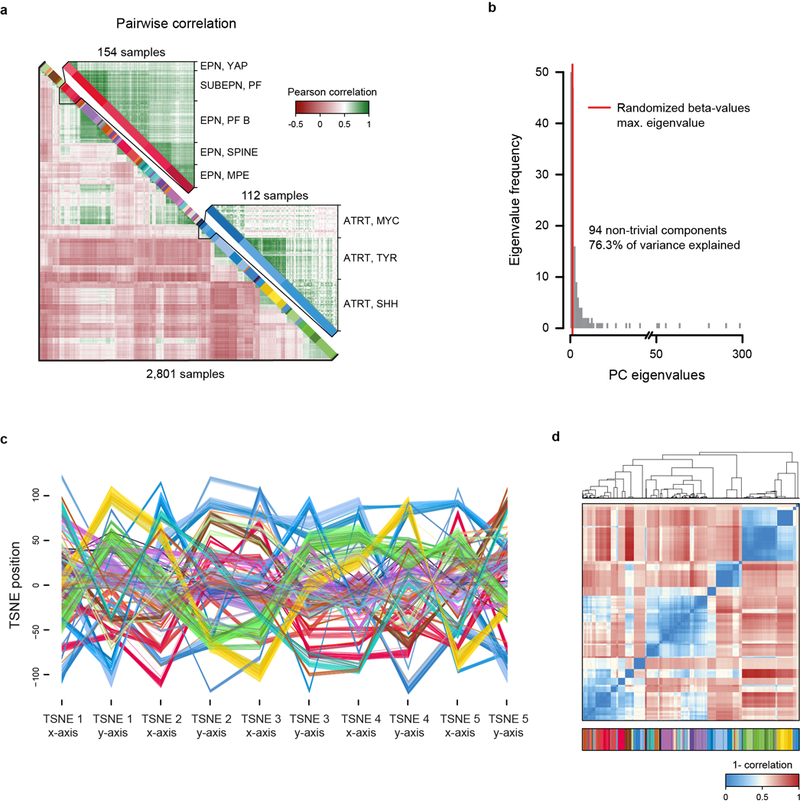
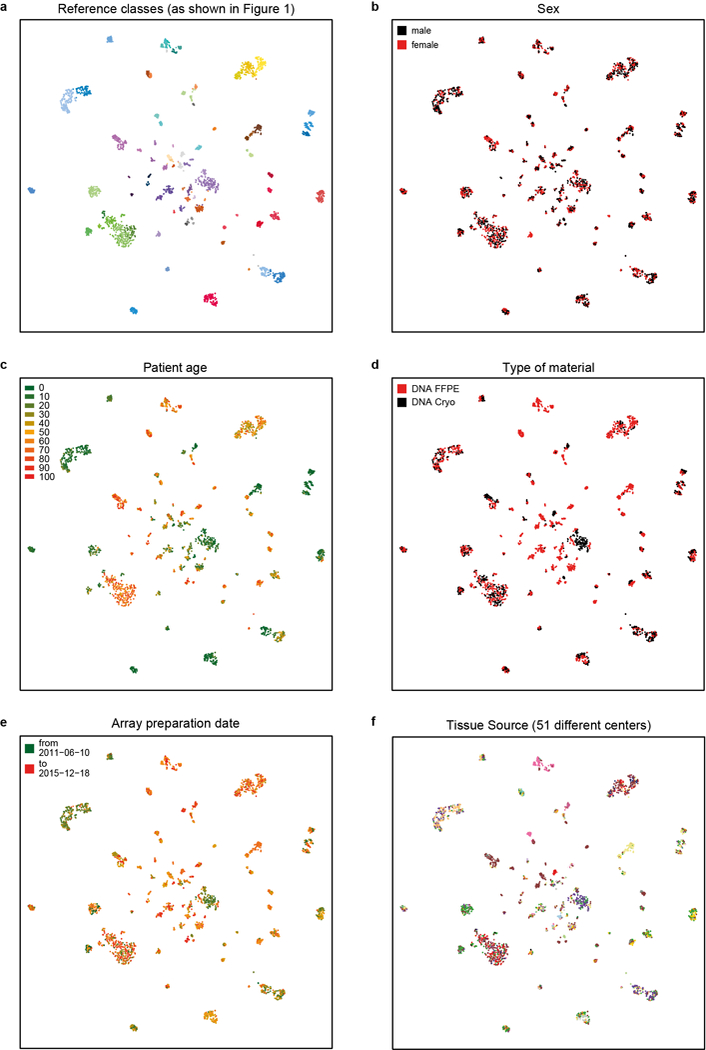
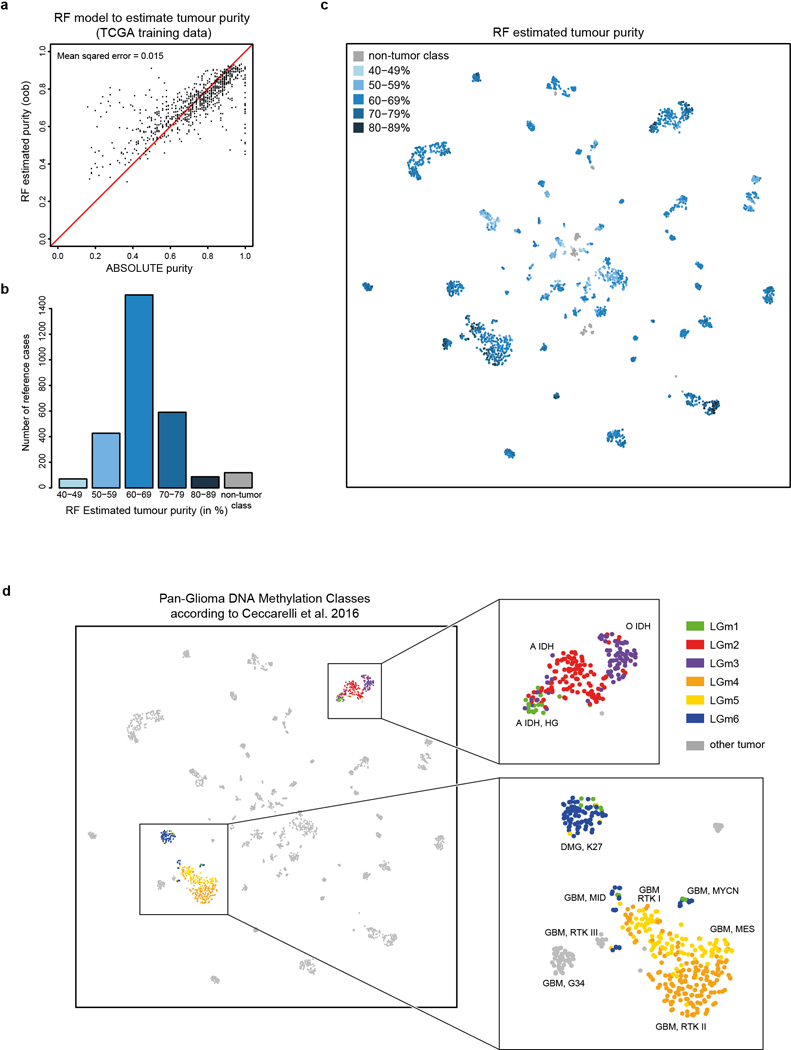
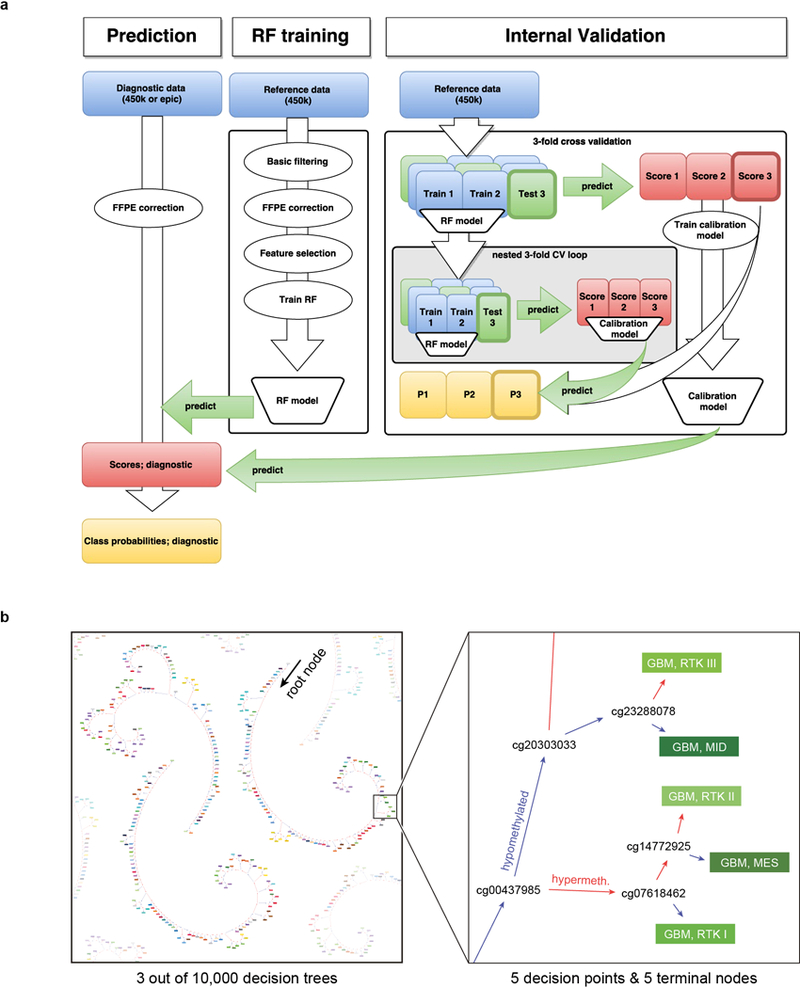
Comment in
-
Machine learning classifies cancer.Nature. 2018 Mar 22;555(7697):446-447. doi: 10.1038/d41586-018-02881-7. Nature. 2018. PMID: 29565394 No abstract available.
-
Machine Learning Improves Diagnosis of CNS Cancers.Cancer Discov. 2018 May;8(5):523-524. doi: 10.1158/2159-8290.CD-NB2018-040. Epub 2018 Mar 30. Cancer Discov. 2018. PMID: 29602827
References
-
- Louis DN, Ohgaki H, Wiestler OD & Cavenee WK WHO Classification of Tumours of the Central Nervous System (revised 4th edition). (IARC, 2016).
Online Only References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
