

Genomic positional conservation identifies topological anchor point RNAs linked to developmental loci
- PMID: 29540241
- PMCID: PMC5853149
- DOI: 10.1186/s13059-018-1405-5
Genomic positional conservation identifies topological anchor point RNAs linked to developmental loci
Abstract
Background: The mammalian genome is transcribed into large numbers of long noncoding RNAs (lncRNAs), but the definition of functional lncRNA groups has proven difficult, partly due to their low sequence conservation and lack of identified shared properties. Here we consider promoter conservation and positional conservation as indicators of functional commonality.
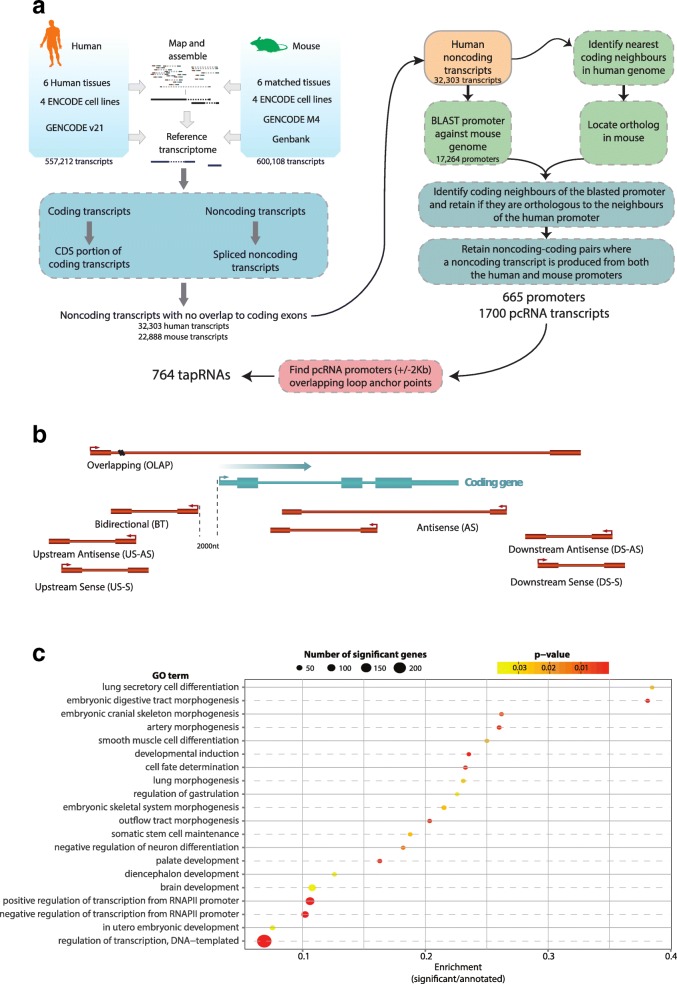
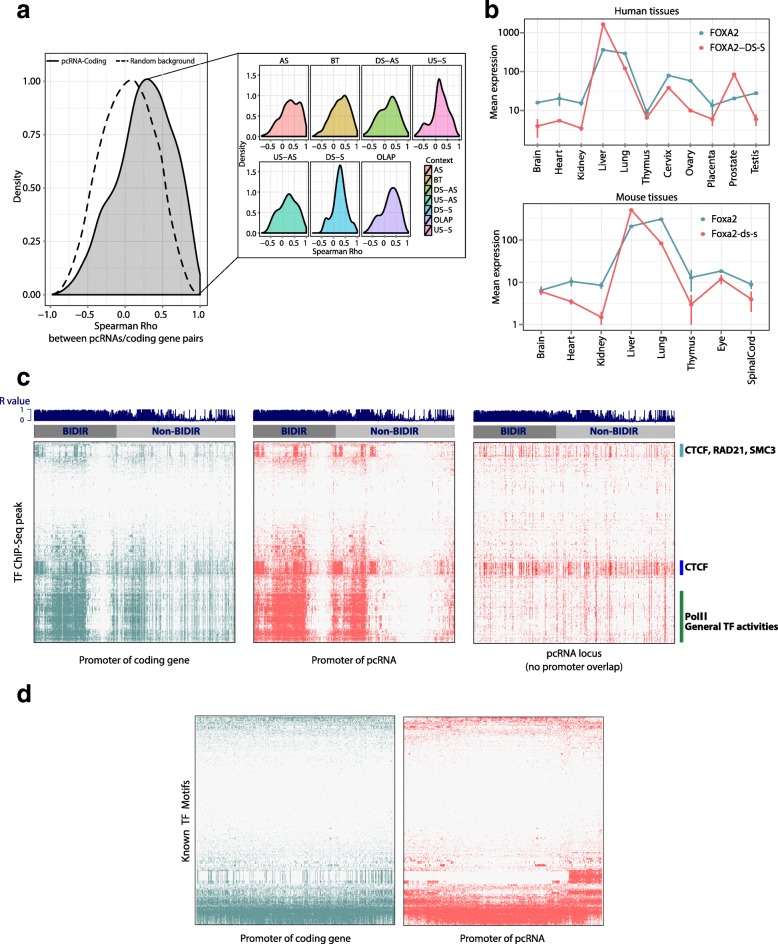
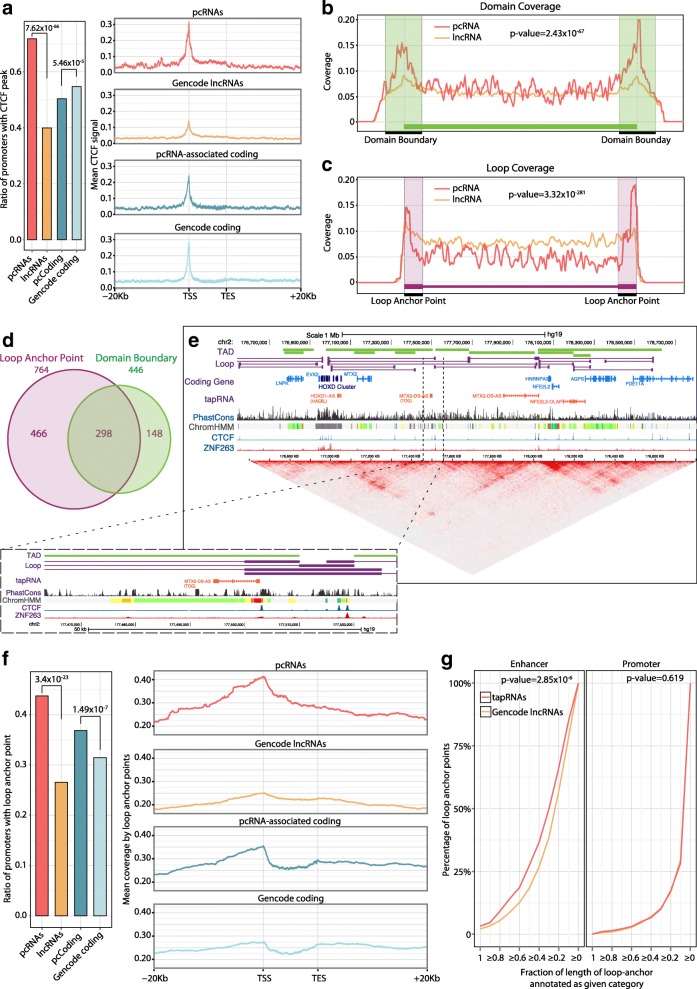
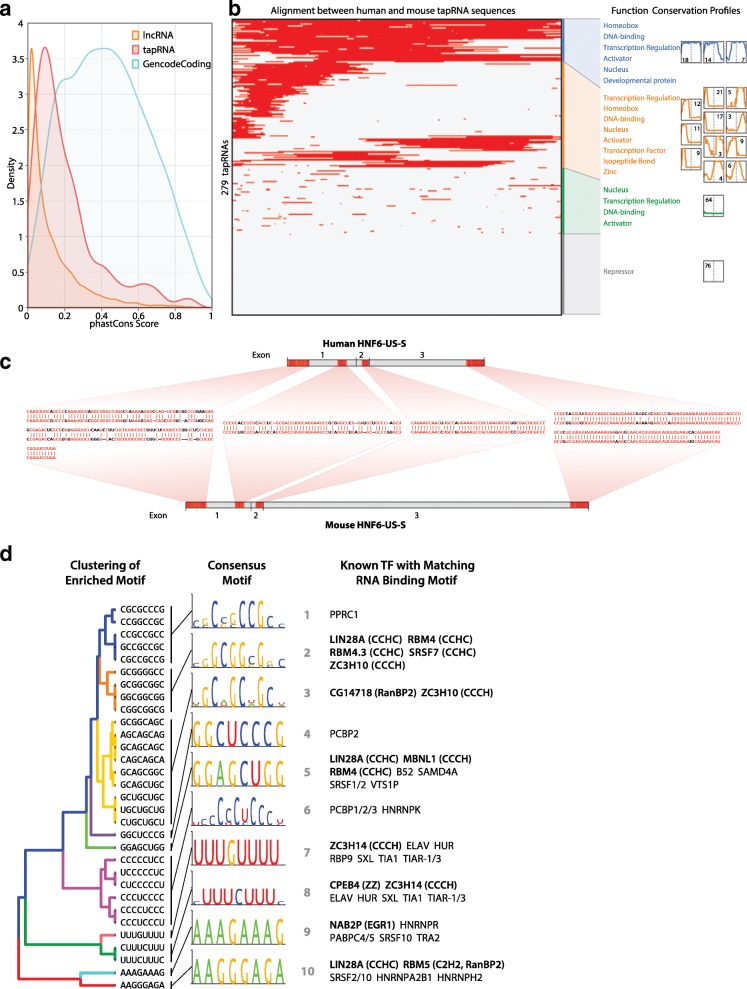
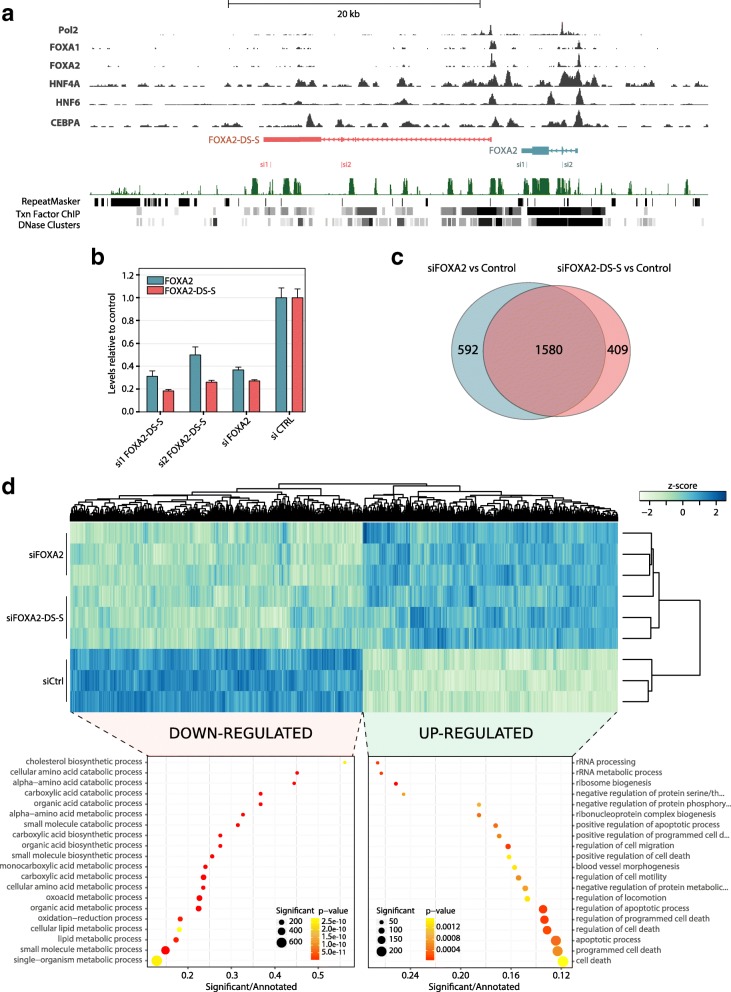
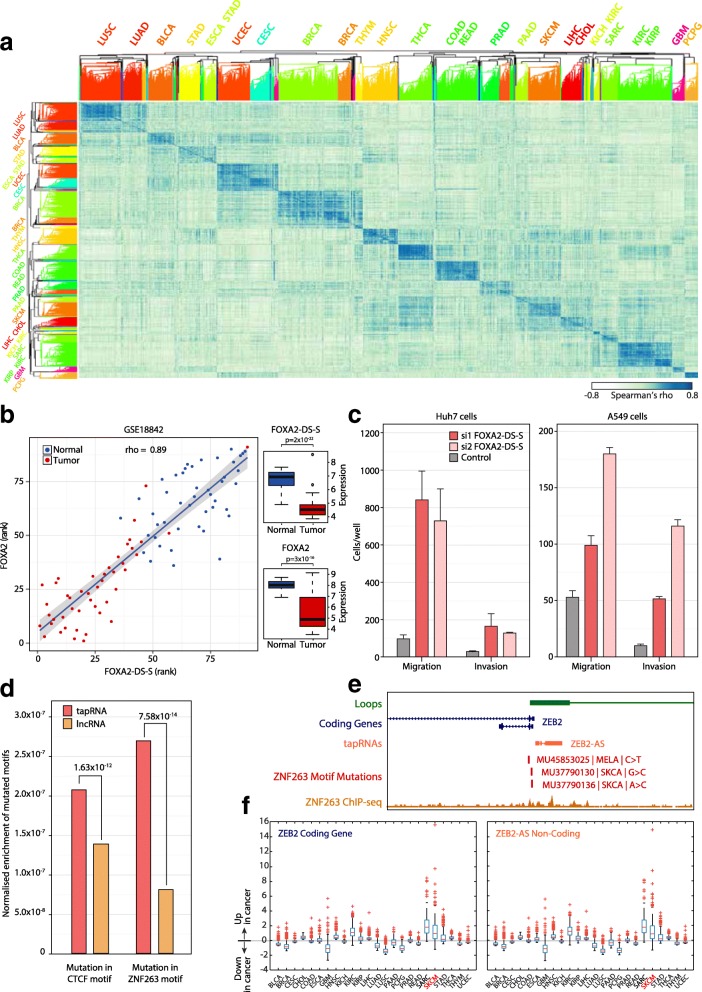
Results: We identify 665 conserved lncRNA promoters in mouse and human that are preserved in genomic position relative to orthologous coding genes. These positionally conserved lncRNA genes are primarily associated with developmental transcription factor loci with which they are coexpressed in a tissue-specific manner. Over half of positionally conserved RNAs in this set are linked to chromatin organization structures, overlapping binding sites for the CTCF chromatin organiser and located at chromatin loop anchor points and borders of topologically associating domains (TADs). We define these RNAs as topological anchor point RNAs (tapRNAs). Characterization of these noncoding RNAs and their associated coding genes shows that they are functionally connected: they regulate each other's expression and influence the metastatic phenotype of cancer cells in vitro in a similar fashion. Furthermore, we find that tapRNAs contain conserved sequence domains that are enriched in motifs for zinc finger domain-containing RNA-binding proteins and transcription factors, whose binding sites are found mutated in cancers.
Conclusions: This work leverages positional conservation to identify lncRNAs with potential importance in genome organization, development and disease. The evidence that many developmental transcription factors are physically and functionally connected to lncRNAs represents an exciting stepping-stone to further our understanding of genome regulation.
Keywords: Cancer; Chromatin architecture; Development; Topology; Zinc finger; lncRNAs.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
