

Osteogenesis imperfecta and therapeutics
- PMID: 29540309
- PMCID: PMC6133774
- DOI: 10.1016/j.matbio.2018.03.010
Osteogenesis imperfecta and therapeutics
Abstract
Osteogenesis imperfecta, or brittle bone disease, is a congenital disease that primarily causes low bone mass and bone fractures but it can negatively affect other organs. It is usually inherited in an autosomal dominant fashion, although rarer recessive and X-chromosome-linked forms of the disease have been identified. In addition to type I collagen, mutations in a number of other genes, often involved in type I collagen synthesis or in the differentiation and function of osteoblasts, have been identified in the last several years. Seldom, the study of a rare disease has delivered such a wealth of new information that have helped our understanding of multiple processes involved in collagen synthesis and bone formation. In this short review I will describe the clinical features and the molecular genetics of the disease, but then focus on how OI dysregulates all aspects of extracellular matrix biology. I will conclude with a discussion about OI therapeutics.
Keywords: Bone; Collagen; Fragility; Genetics; Mineralization; Osteogenesis imperfecta; Proteoglycans.
Copyright © 2018 Elsevier B.V. All rights reserved.
Figures
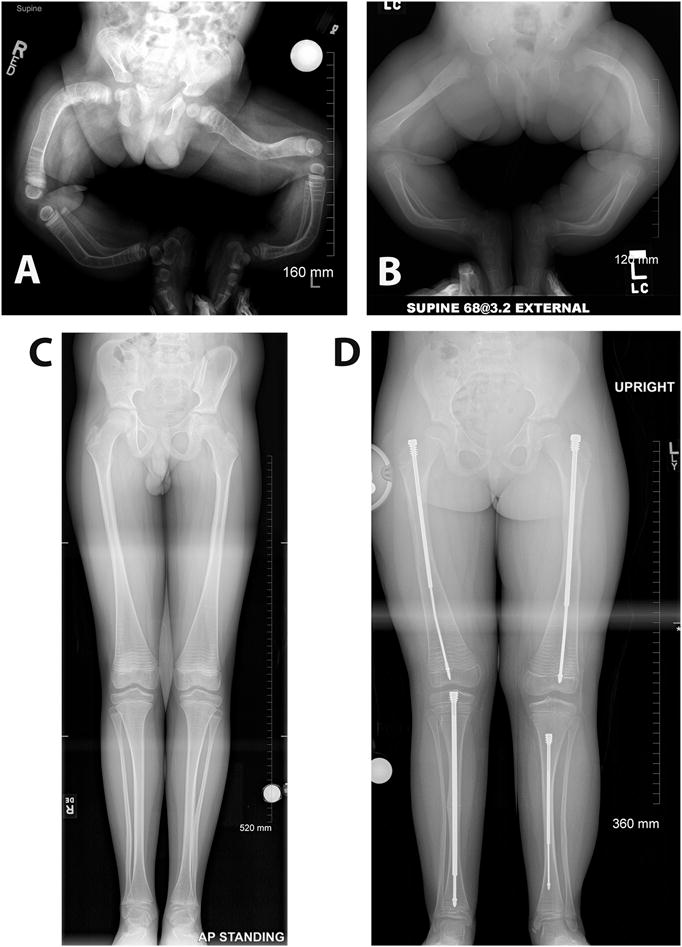
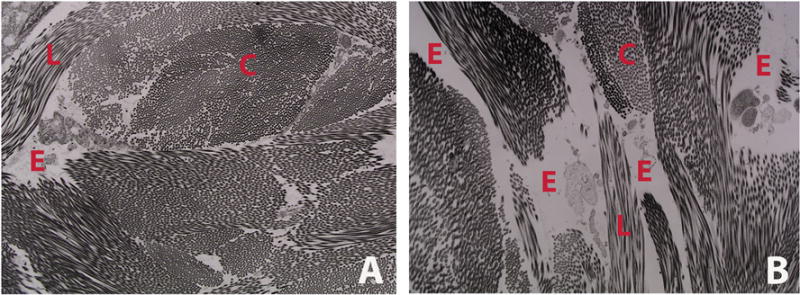
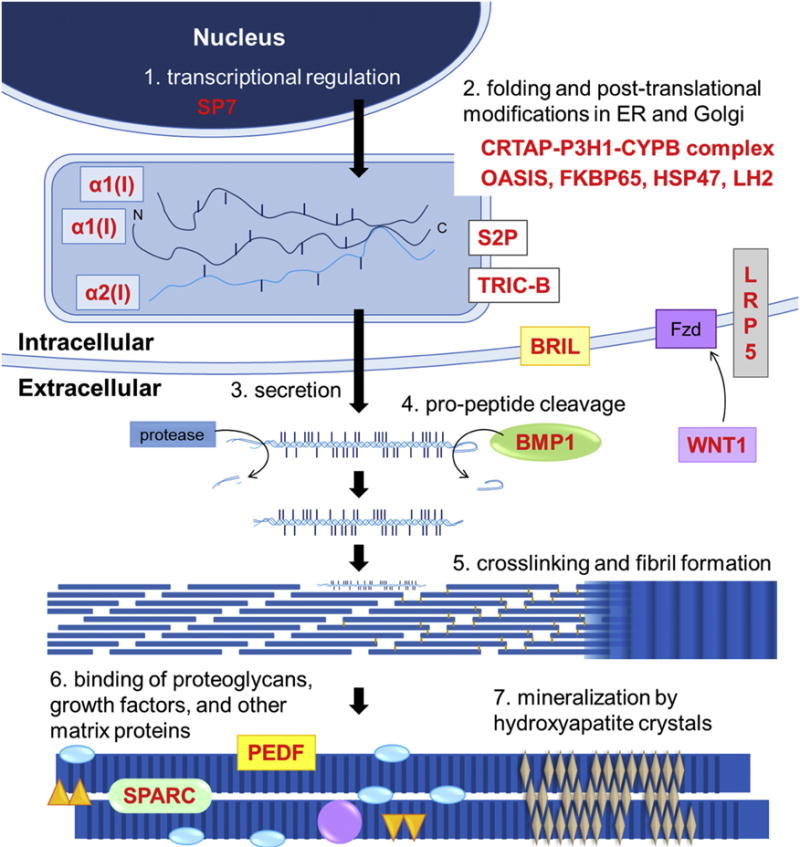
References
-
- Vrolik W. Tabulae ad illustrandam embryogenesin hominis et mammalium, naturalem tam abnormem 1849
-
- Baljet B. Aspects of the history of osteogenesis imperfecta (Vrolik’s syndrome) Ann Anat. 2002;184:1–7. - PubMed
-
- Seedorf K. Osteogenesis imperfecta: a study of clinical features and heredity based on 55 Danish families comprising 180 affected members. Copenhagen: UniversitetsforlagetI Aarhus; 1949.
-
- Smars G. Clinical, Genetic, Epidemiological and Socio-medical Aspects. Stockholm: Scandinavian University Books; 1961. Osteogenesis imperfecta in Sweden.
-
- Sillence DO, Rimoin DL. Classification of osteogenesis imperfect. Lancet. 1978;1:1041–1042. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
