

Compression force sensing regulates integrin αIIbβ3 adhesive function on diabetic platelets
- PMID: 29540687
- PMCID: PMC5852038
- DOI: 10.1038/s41467-018-03430-6
Compression force sensing regulates integrin αIIbβ3 adhesive function on diabetic platelets
Abstract
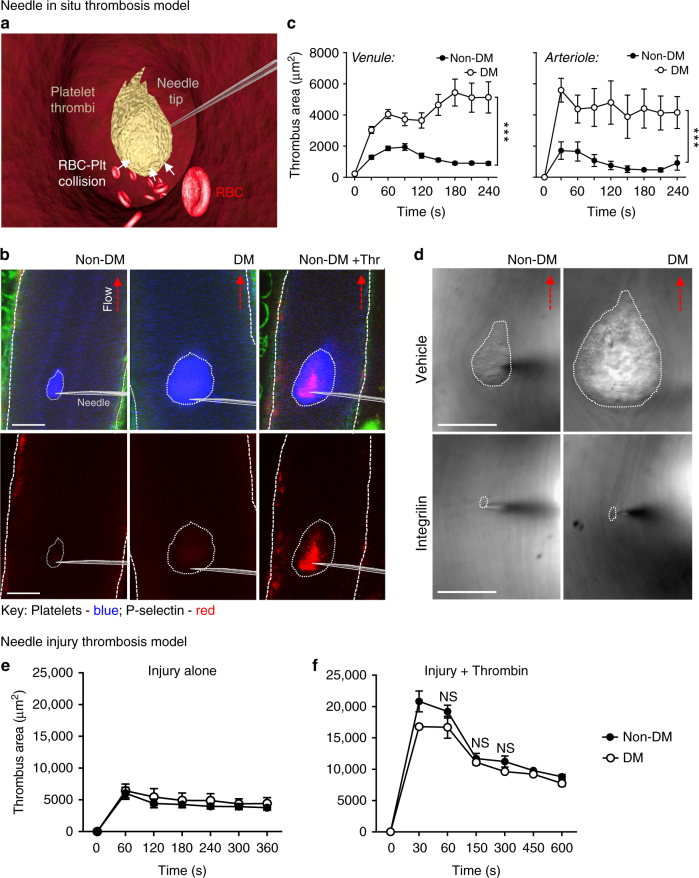
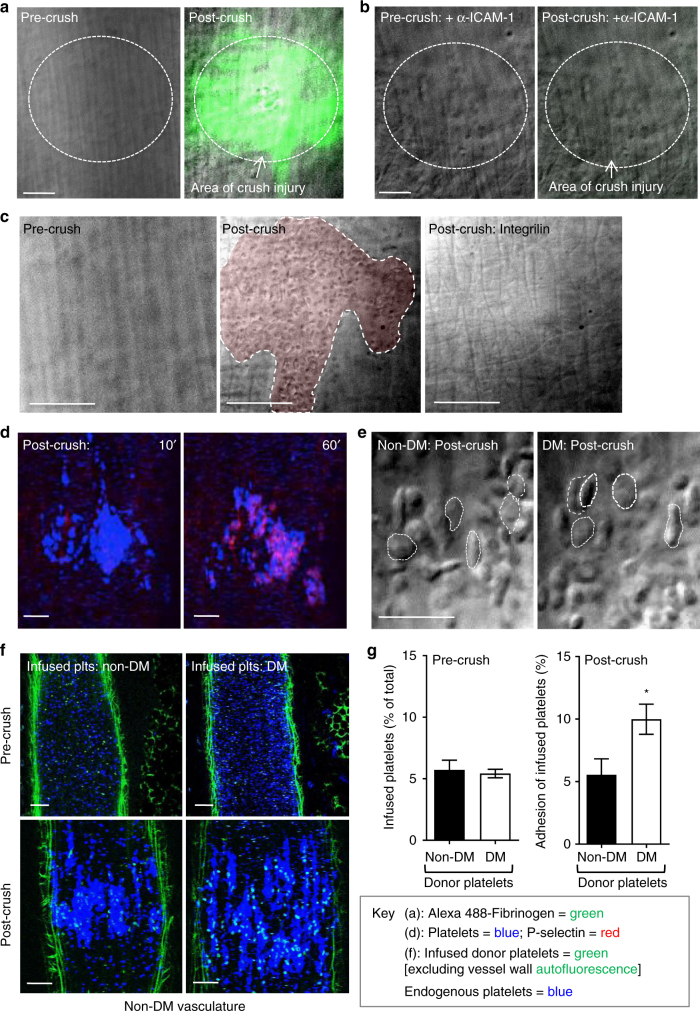
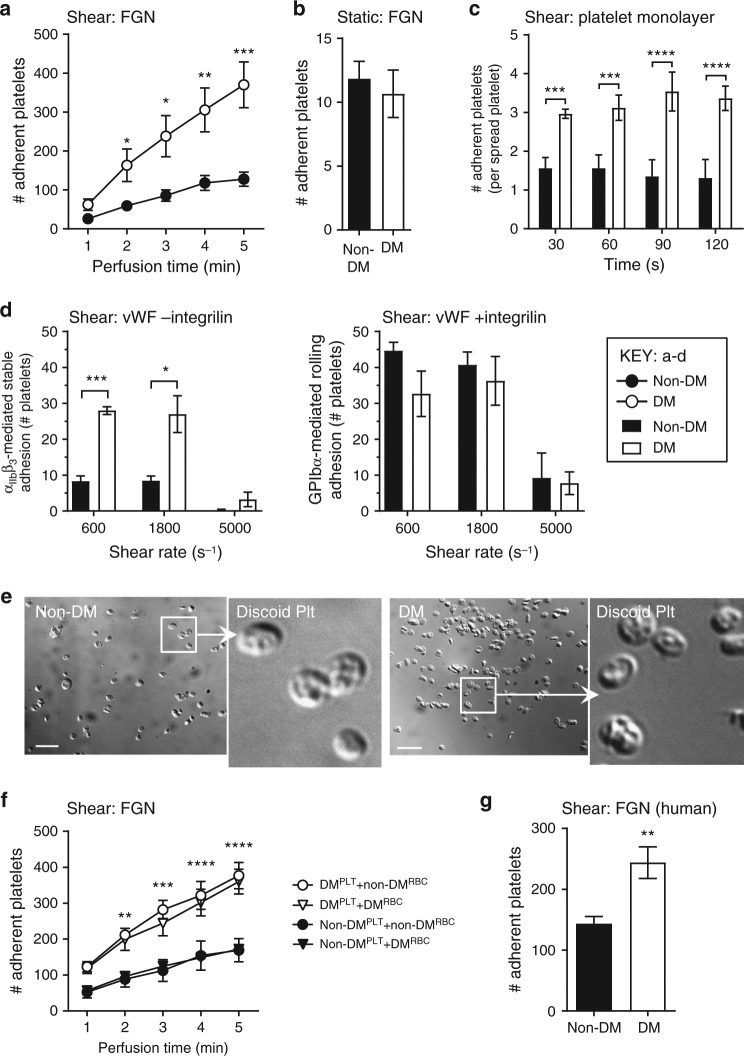
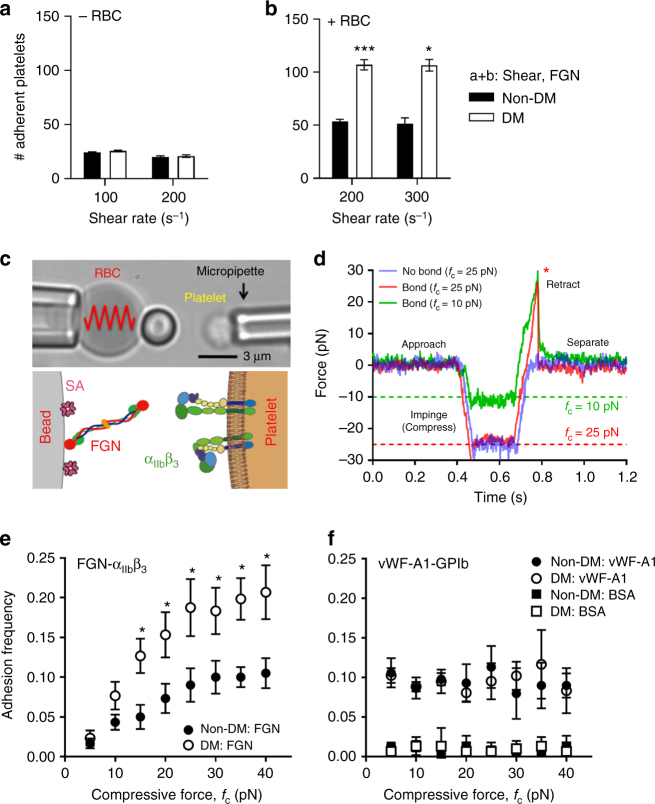
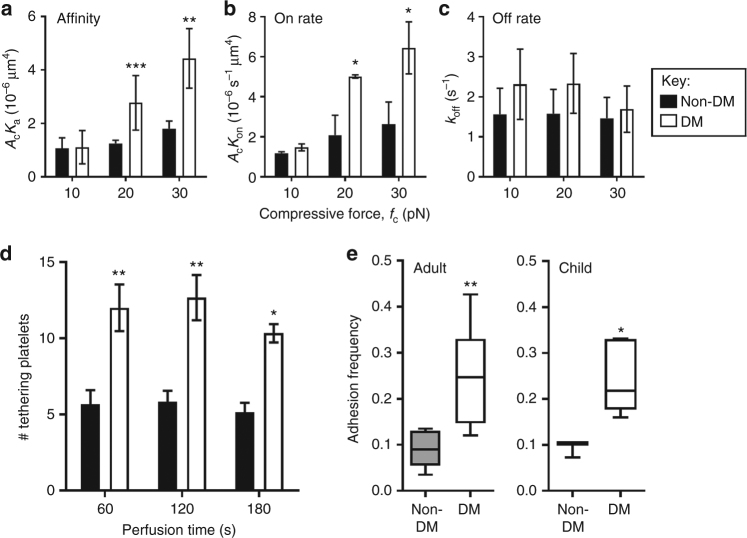
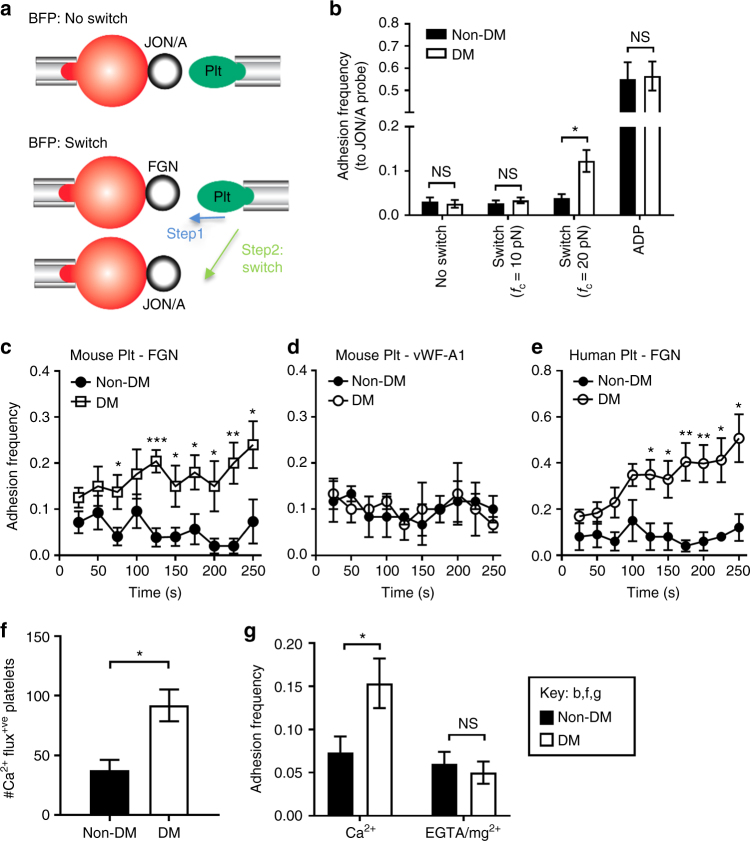
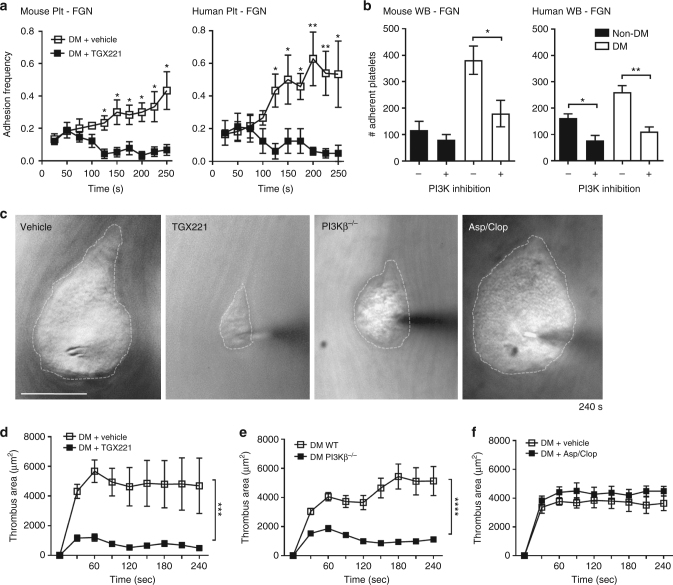
Diabetes is associated with an exaggerated platelet thrombotic response at sites of vascular injury. Biomechanical forces regulate platelet activation, although the impact of diabetes on this process remains ill-defined. Using a biomembrane force probe (BFP), we demonstrate that compressive force activates integrin αIIbβ3 on discoid diabetic platelets, increasing its association rate with immobilized fibrinogen. This compressive force-induced integrin activation is calcium and PI 3-kinase dependent, resulting in enhanced integrin affinity maturation and exaggerated shear-dependent platelet adhesion. Analysis of discoid platelet aggregation in the mesenteric circulation of mice confirmed that diabetes leads to a marked enhancement in the formation and stability of discoid platelet aggregates, via a mechanism that is not inhibited by therapeutic doses of aspirin and clopidogrel, but is eliminated by PI 3-kinase inhibition. These studies demonstrate the existence of a compression force sensing mechanism linked to αIIbβ3 adhesive function that leads to a distinct prothrombotic phenotype in diabetes.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
