

Prediction and interpretation of deleterious coding variants in terms of protein structural stability
- PMID: 29540703
- PMCID: PMC5852127
- DOI: 10.1038/s41598-018-22531-2
Prediction and interpretation of deleterious coding variants in terms of protein structural stability
Abstract
The classification of human genetic variants into deleterious and neutral is a challenging issue, whose complexity is rooted in the large variety of biophysical mechanisms that can be responsible for disease conditions. For non-synonymous mutations in structured proteins, one of these is the protein stability change, which can lead to loss of protein structure or function. We developed a stability-driven knowledge-based classifier that uses protein structure, artificial neural networks and solvent accessibility-dependent combinations of statistical potentials to predict whether destabilizing or stabilizing mutations are disease-causing. Our predictor yields a balanced accuracy of 71% in cross validation. As expected, it has a very high positive predictive value of 89%: it predicts with high accuracy the subset of mutations that are deleterious because of stability issues, but is by construction unable of classifying variants that are deleterious for other reasons. Its combination with an evolutionary-based predictor increases the balanced accuracy up to 75%, and allowed predicting more than 1/4 of the variants with 95% positive predictive value. Our method, called SNPMuSiC, can be used with both experimental and modeled structures and compares favorably with other prediction tools on several independent test sets. It constitutes a step towards interpreting variant effects at the molecular scale. SNPMuSiC is freely available at https://soft.dezyme.com/ .
Conflict of interest statement
The authors declare no competing interests.
Figures

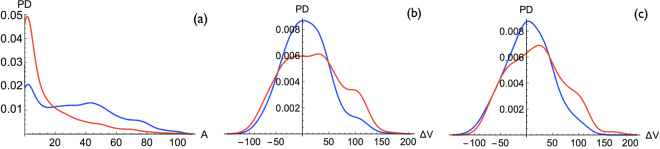

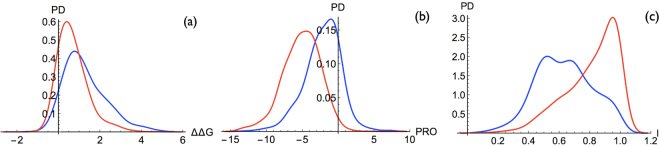
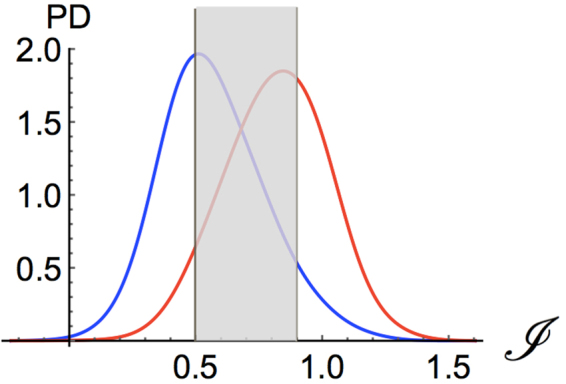
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
