

Kir6.1/K-ATP channel modulates microglia phenotypes: implication in Parkinson's disease
- PMID: 29540778
- PMCID: PMC5852118
- DOI: 10.1038/s41419-018-0437-9
Kir6.1/K-ATP channel modulates microglia phenotypes: implication in Parkinson's disease
Abstract
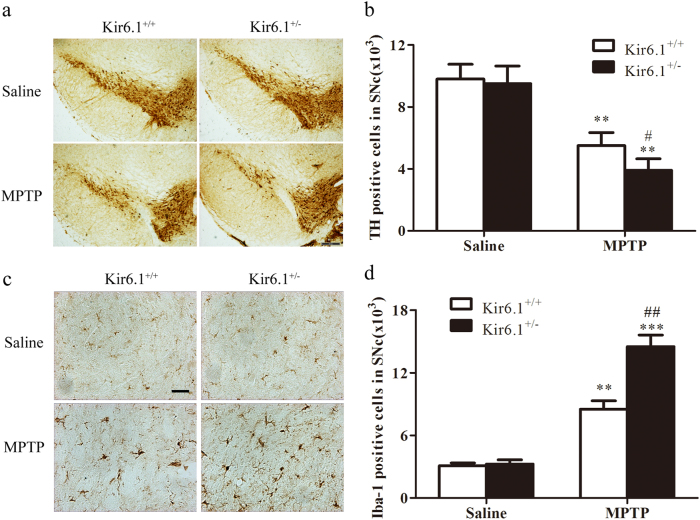
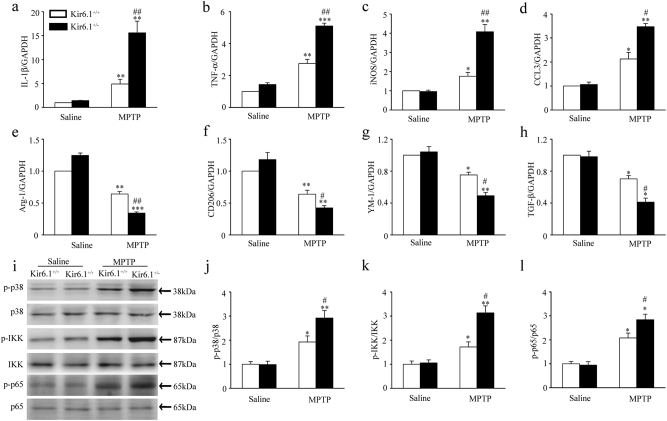
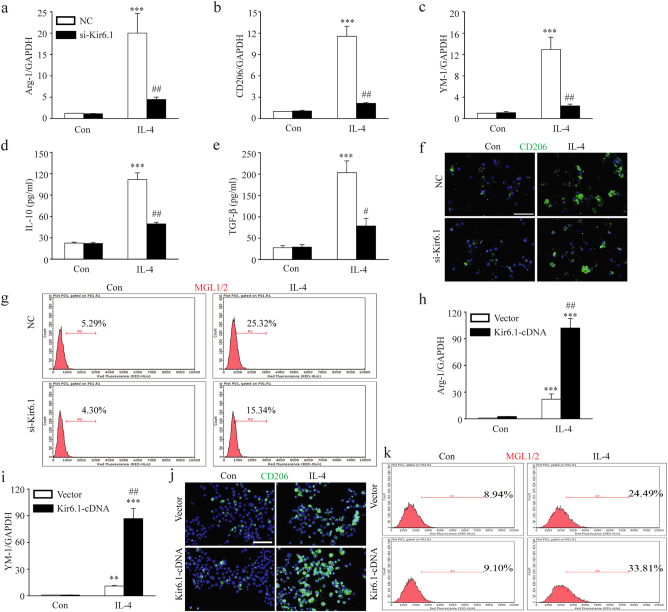
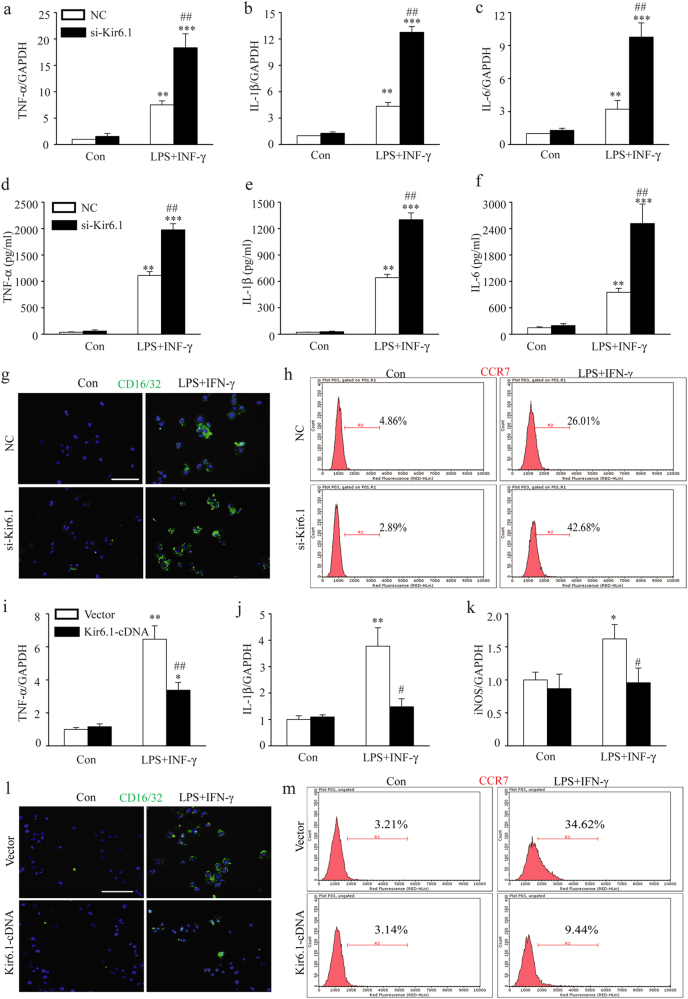
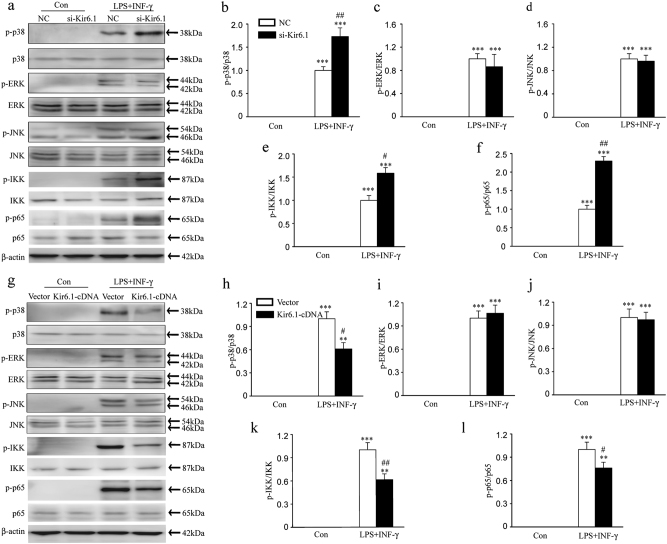
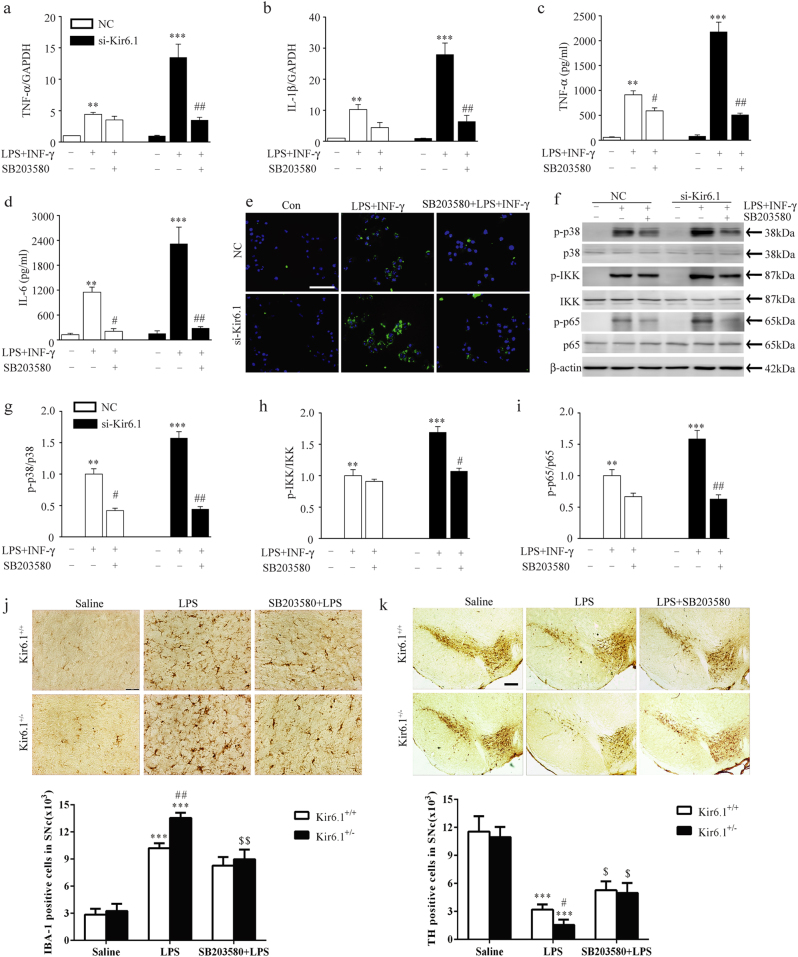
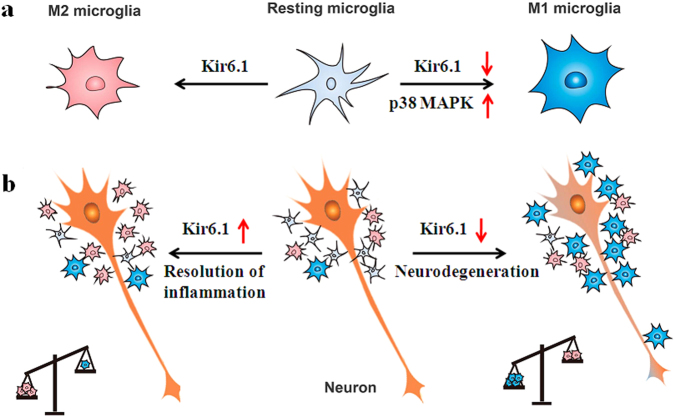
Classical activation (M1 phenotype) and alternative activation (M2 phenotype) are the two polars of microglial activation states that can produce either neurotoxic or neuroprotective effects in the immune pathogenesis of Parkinson's disease (PD). Exploiting the beneficial properties of microglia cells by modulating their polarization states provides great potential for the treatment of PD. However, the mechanism that regulates microglia polarization remains elusive. Here we demonstrated that Kir6.1-containing ATP-sensitive potassium (Kir6.1/K-ATP) channel switched microglia from the detrimental M1 phenotype toward the beneficial M2 phenotype. Kir6.1 knockdown inhibited M2 polarization and simultaneously exaggerated M1 microglial inflammatory responses, while Kir6.1 overexpression promoted M2 polarization and synchronously alleviated the toxic phase of M1 microglia polarization. Furthermore, we observed that the Kir6.1 deficiency dramatically exacerbated dopaminergic neuron death companied by microglia activation in mouse model of PD. Mechanistically, Kir6.1 deficiency enhanced the activation of p38 MAPK-NF-κB pathway and increased the ratio of M1/M2 markers in the substantia nigra compacta of mouse model of PD. Suppression of p38 MAPK in vivo partially rescued the deleterious effects of Kir6.1 ablation on microglia phenotype and dopaminergic neuron death. Collectively, our findings reveal that Kir6.1/K-ATP channel modulates microglia phenotypes transition via inhibition of p38 MAPK-NF-κB signaling pathway and Kir6.1/K-ATP channel may be a promising therapeutic target for PD.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Kir6.1/K-ATP channel on astrocytes protects against dopaminergic neurodegeneration in the MPTP mouse model of Parkinson's disease via promoting mitophagy.Brain Behav Immun. 2019 Oct;81:509-522. doi: 10.1016/j.bbi.2019.07.009. Epub 2019 Jul 6. Brain Behav Immun. 2019. PMID: 31288070
-
Spermidine Inhibits M1 Microglia Polarization in a Mouse Model of Parkinson's Disease and BV2 Cells via NF-κB/STAT-1 Pathway.Brain Behav. 2025 Mar;15(3):e70410. doi: 10.1002/brb3.70410. Brain Behav. 2025. PMID: 40059454 Free PMC article.
-
Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson's disease.Cell Death Differ. 2014 Mar;21(3):369-80. doi: 10.1038/cdd.2013.159. Epub 2013 Nov 8. Cell Death Differ. 2014. PMID: 24212761 Free PMC article.
-
Therapeutic potential of ATP-sensitive potassium channels in Parkinson's disease.Brain Res Bull. 2021 Apr;169:1-7. doi: 10.1016/j.brainresbull.2021.01.003. Epub 2021 Jan 9. Brain Res Bull. 2021. PMID: 33434622 Review.
-
Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson's Disease.Front Aging Neurosci. 2017 Jun 8;9:176. doi: 10.3389/fnagi.2017.00176. eCollection 2017. Front Aging Neurosci. 2017. PMID: 28642697 Free PMC article. Review.
Cited by
-
The activation impact of lactobacillus-derived extracellular vesicles on lipopolysaccharide-induced microglial cell.BMC Microbiol. 2024 Feb 28;24(1):70. doi: 10.1186/s12866-024-03217-4. BMC Microbiol. 2024. PMID: 38418961 Free PMC article.
-
Microglia in Alzheimer's disease: pathogenesis, mechanisms, and therapeutic potentials.Front Aging Neurosci. 2023 Jun 15;15:1201982. doi: 10.3389/fnagi.2023.1201982. eCollection 2023. Front Aging Neurosci. 2023. PMID: 37396657 Free PMC article. Review.
-
Potassium and calcium channels in different nerve cells act as therapeutic targets in neurological disorders.Neural Regen Res. 2025 May 1;20(5):1258-1276. doi: 10.4103/NRR.NRR-D-23-01766. Epub 2024 Jun 3. Neural Regen Res. 2025. PMID: 38845230 Free PMC article.
-
The dual face of microglia (M1/M2) as a potential target in the protective effect of nutraceuticals against neurodegenerative diseases.Front Aging. 2023 Sep 6;4:1231706. doi: 10.3389/fragi.2023.1231706. eCollection 2023. Front Aging. 2023. PMID: 37744008 Free PMC article. Review.
-
Substantia nigra Smad3 signaling deficiency: relevance to aging and Parkinson's disease and roles of microglia, proinflammatory factors, and MAPK.J Neuroinflammation. 2020 Nov 16;17(1):342. doi: 10.1186/s12974-020-02023-9. J Neuroinflammation. 2020. PMID: 33198771 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
