

Cancer cell metabolic plasticity allows resistance to NAMPT inhibition but invariably induces dependence on LDHA
- PMID: 29541451
- PMCID: PMC5844108
- DOI: 10.1186/s40170-018-0174-7
Cancer cell metabolic plasticity allows resistance to NAMPT inhibition but invariably induces dependence on LDHA
Abstract
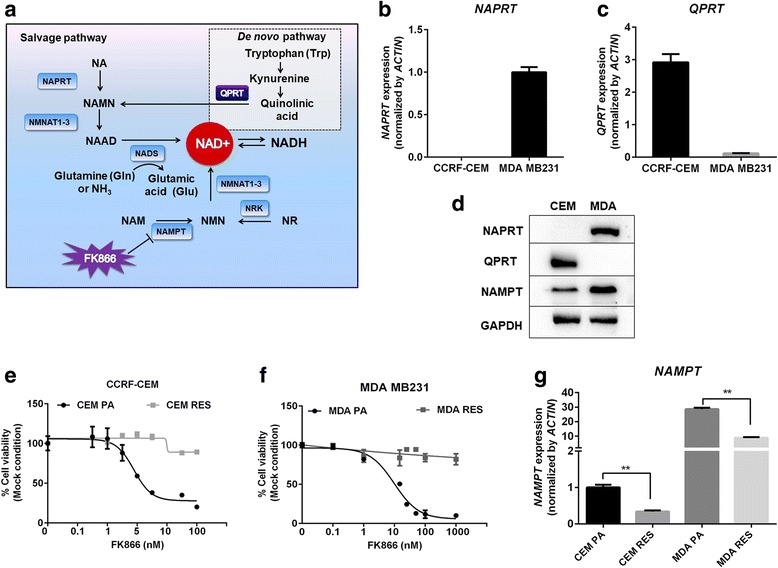
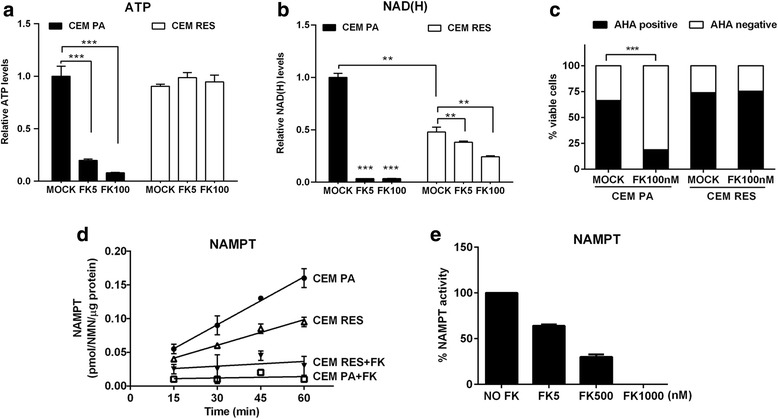
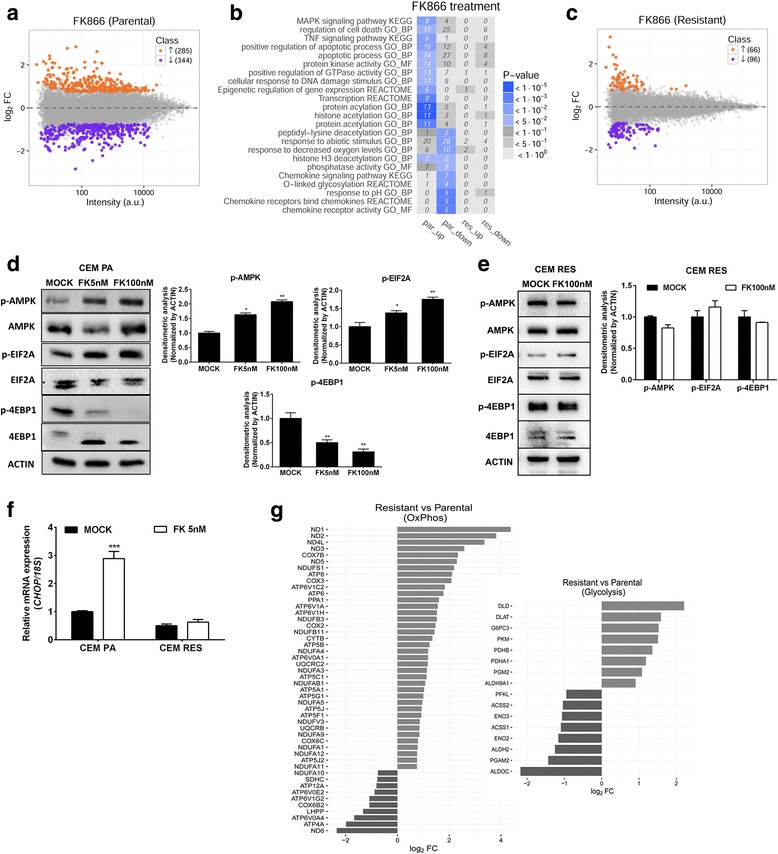
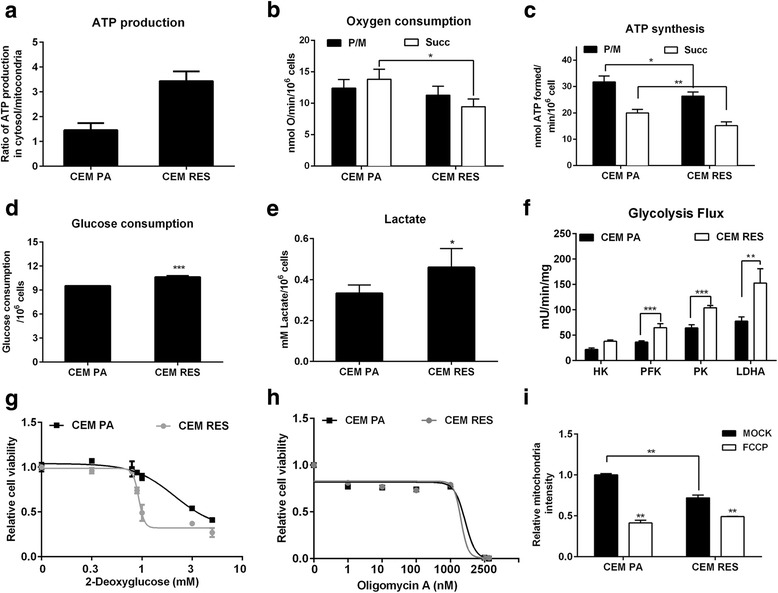
Background: Inhibitors of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in NAD+ biosynthesis from nicotinamide, exhibit anticancer effects in preclinical models. However, continuous exposure to NAMPT inhibitors, such as FK866, can induce acquired resistance.
Methods: We developed FK866-resistant CCRF-CEM (T cell acute lymphoblastic leukemia) and MDA MB231 (breast cancer) models, and by exploiting an integrated approach based on genetic, biochemical, and genome wide analyses, we annotated the drug resistance mechanisms.
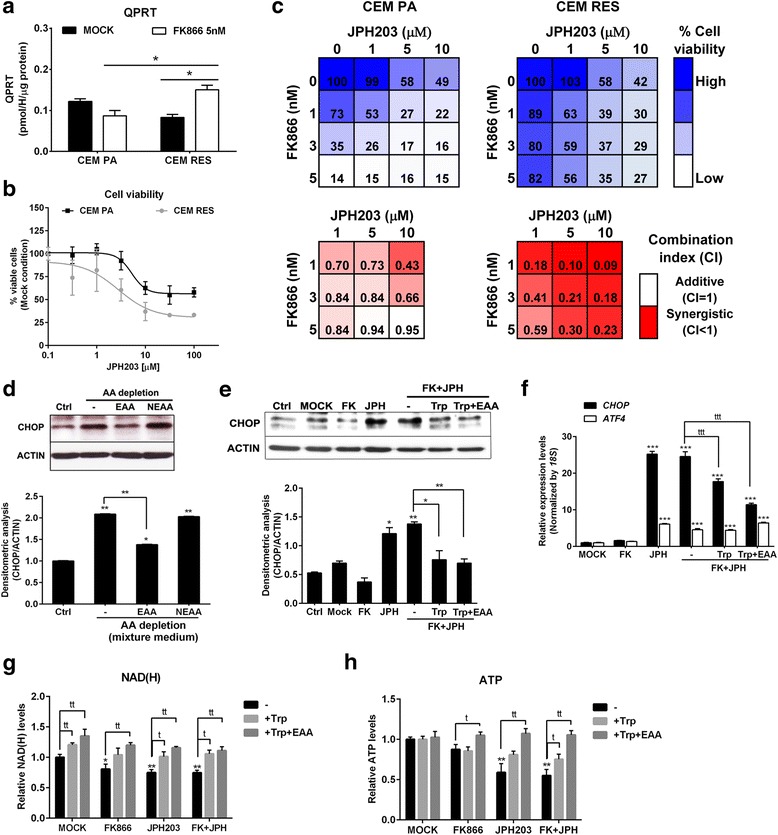
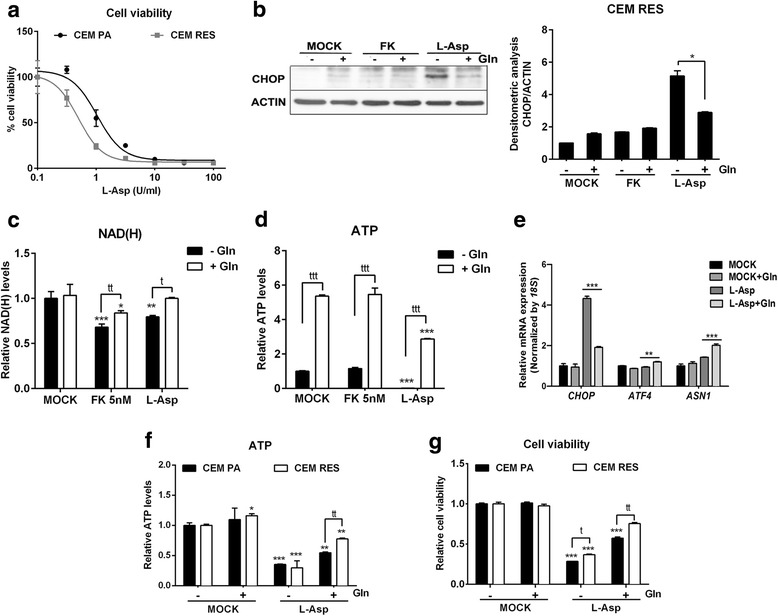
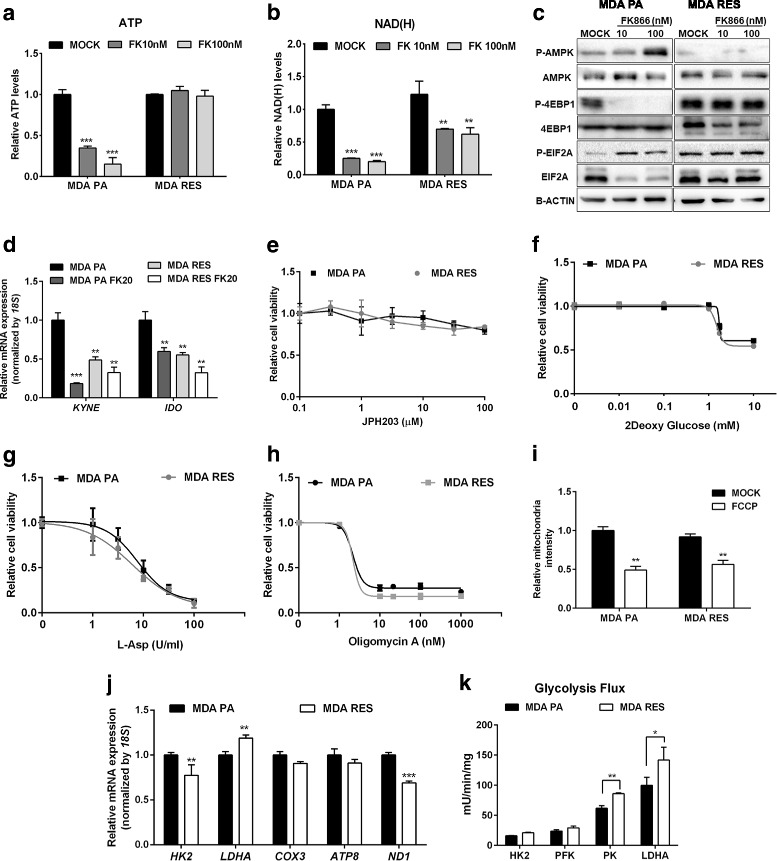
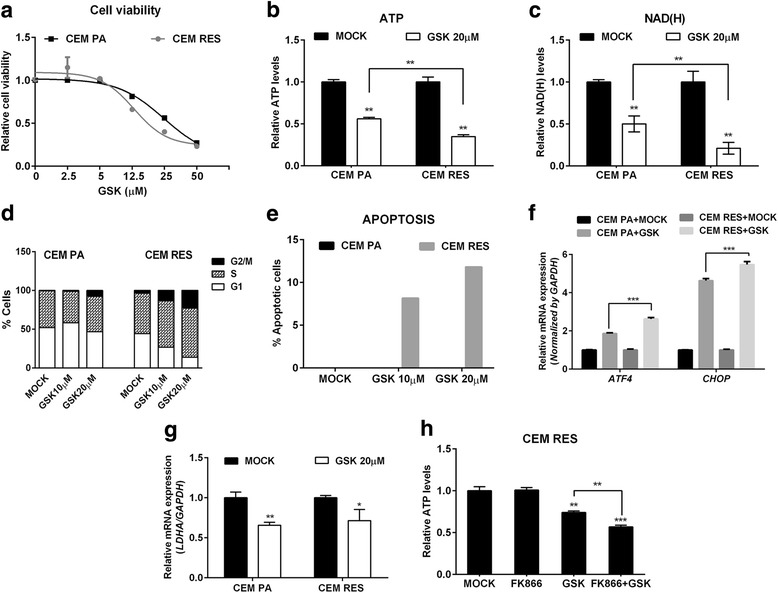
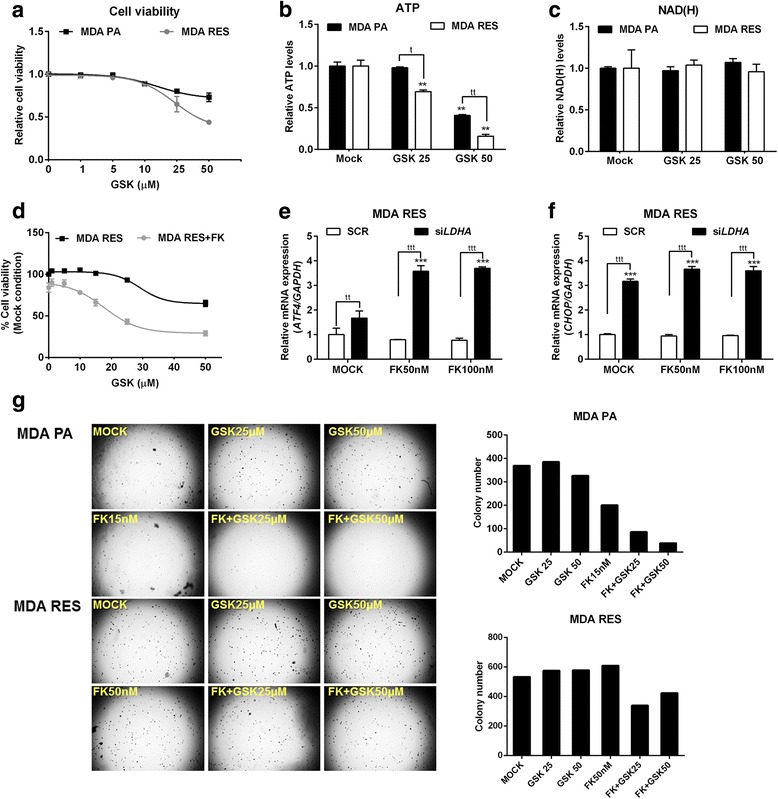
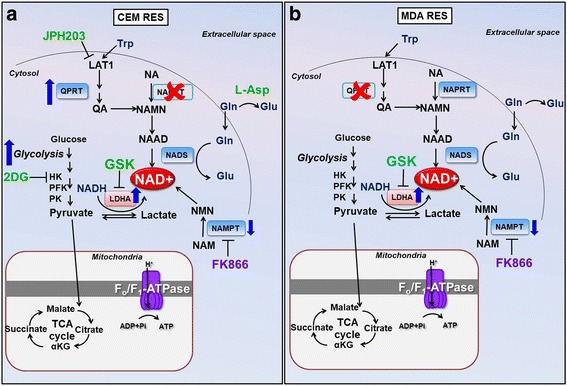
Results: Acquired resistance to FK866 was independent of NAMPT mutations but rather was based on a shift towards a glycolytic metabolism and on lactate dehydrogenase A (LDHA) activity. In addition, resistant CCRF-CEM cells, which exhibit high quinolinate phosphoribosyltransferase (QPRT) activity, also exploited amino acid catabolism as an alternative source for NAD+ production, becoming addicted to tryptophan and glutamine and sensitive to treatment with the amino acid transport inhibitor JPH203 and with l-asparaginase, which affects glutamine exploitation. Vice versa, in line with their low QPRT expression, FK866-resistant MDA MB231 did not rely on amino acids for their resistance phenotype.
Conclusions: Our study identifies novel mechanisms of resistance to NAMPT inhibition, which may be useful to design more rational strategies for targeting cancer metabolism.
Keywords: Amino acid metabolism; Drug resistance; LDHA; NAMPT; QPRT.
Conflict of interest statement
Not applicable.Not applicable.The authors declare that they have no competing interests.Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
