

Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data
- PMID: 29544553
- PMCID: PMC5856372
- DOI: 10.1186/s13059-018-1408-2
Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data
Erratum in
-
Correction to: Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data.Genome Biol. 2019 Feb 22;20(1):43. doi: 10.1186/s13059-019-1656-9. Genome Biol. 2019. PMID: 30795792 Free PMC article.
Abstract
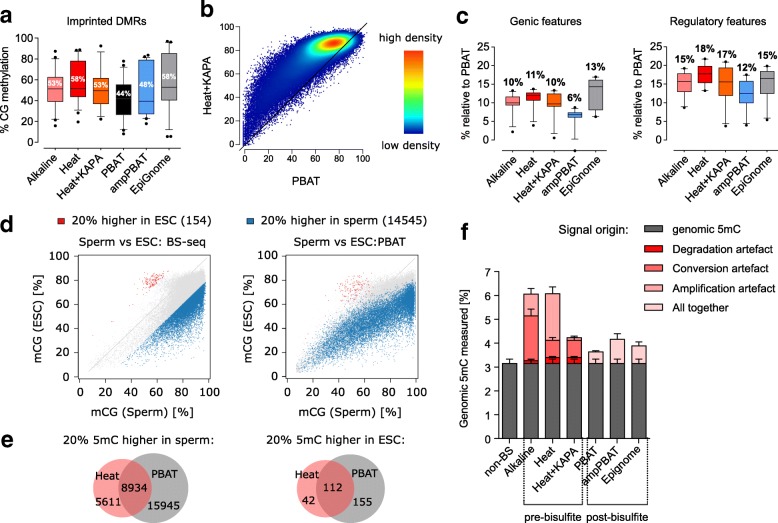
Background: Whole-genome bisulfite sequencing (WGBS) is becoming an increasingly accessible technique, used widely for both fundamental and disease-oriented research. Library preparation methods benefit from a variety of available kits, polymerases and bisulfite conversion protocols. Although some steps in the procedure, such as PCR amplification, are known to introduce biases, a systematic evaluation of biases in WGBS strategies is missing.
Results: We perform a comparative analysis of several commonly used pre- and post-bisulfite WGBS library preparation protocols for their performance and quality of sequencing outputs. Our results show that bisulfite conversion per se is the main trigger of pronounced sequencing biases, and PCR amplification builds on these underlying artefacts. The majority of standard library preparation methods yield a significantly biased sequence output and overestimate global methylation. Importantly, both absolute and relative methylation levels at specific genomic regions vary substantially between methods, with clear implications for DNA methylation studies.
Conclusions: We show that amplification-free library preparation is the least biased approach for WGBS. In protocols with amplification, the choice of bisulfite conversion protocol or polymerase can significantly minimize artefacts. To aid with the quality assessment of existing WGBS datasets, we have integrated a bias diagnostic tool in the Bismark package and offer several approaches for consideration during the preparation and analysis of WGBS datasets.
Keywords: Artefacts; Biases; Bisulfite conversion; DNA methylation; Degradation; GC skew; NGS; Polymerase; WGBS.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing financial interests. WR is a consultant and shareholder at Cambridge Epigenetix Ltd.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
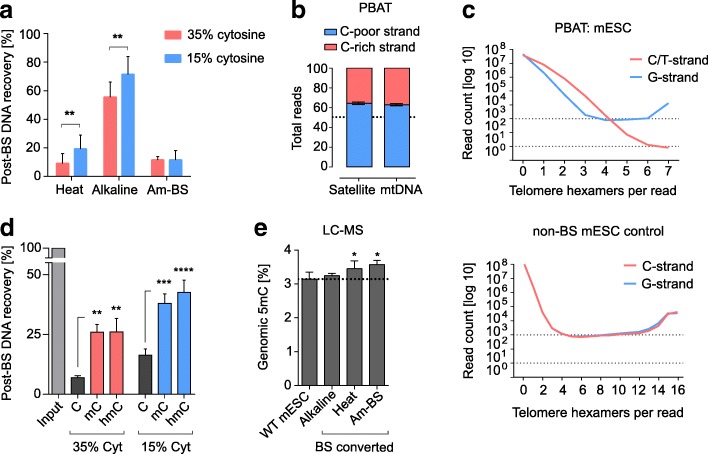
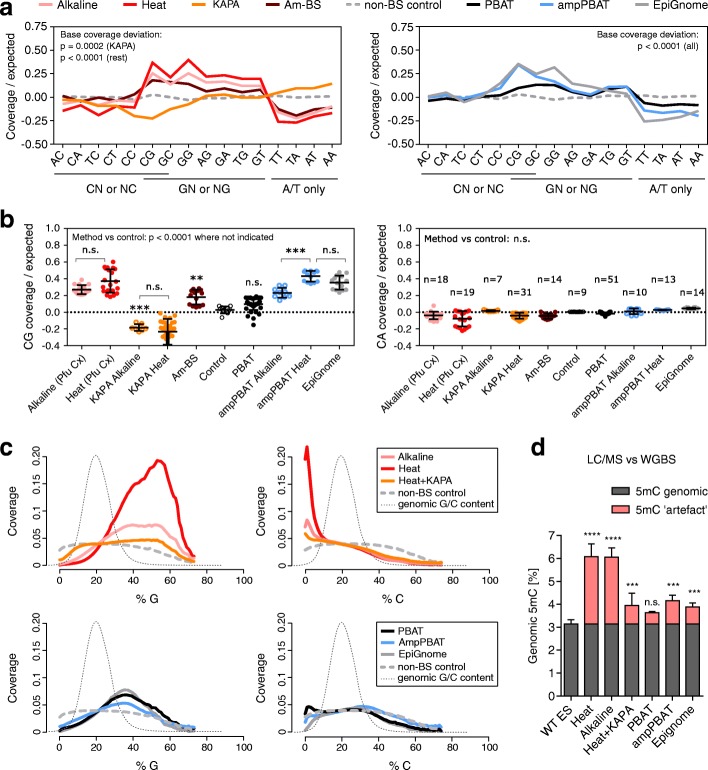
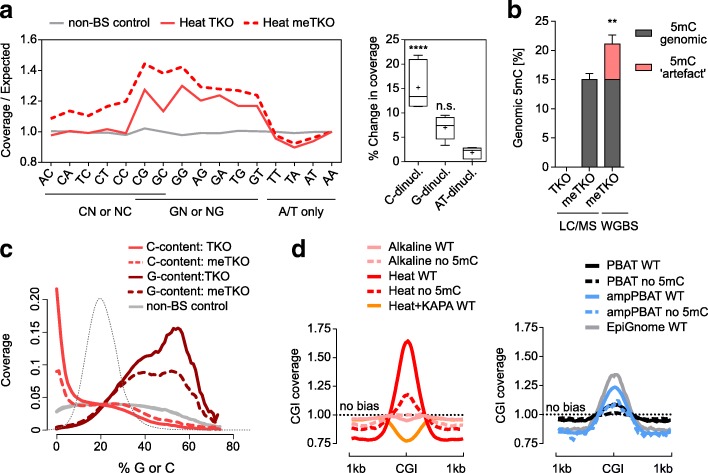
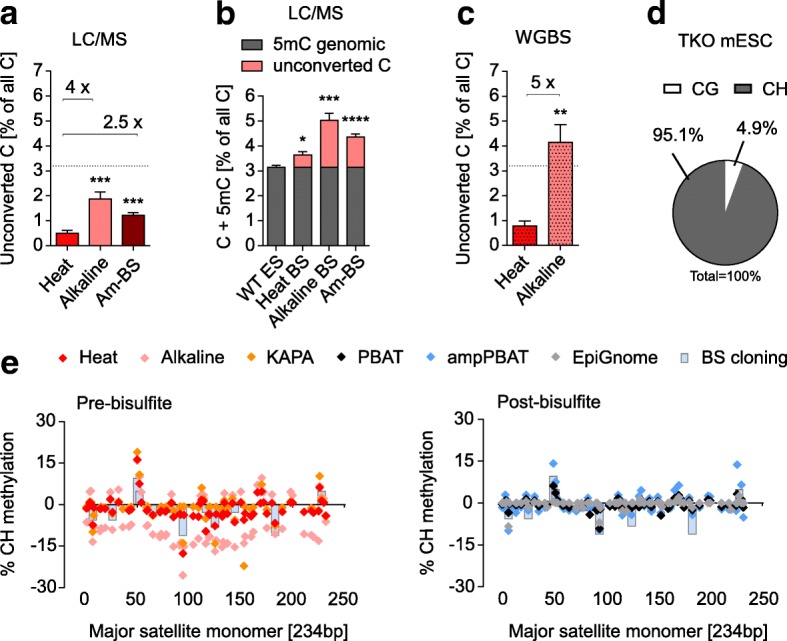

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
