

Epigenetic control of gene regulation during development and disease: A view from the retina
- PMID: 29544768
- PMCID: PMC6054546
- DOI: 10.1016/j.preteyeres.2018.03.002
Epigenetic control of gene regulation during development and disease: A view from the retina
Abstract
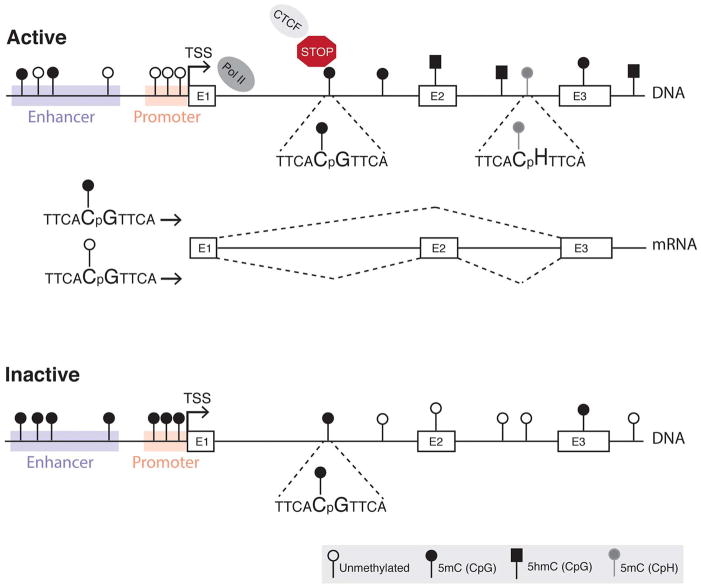
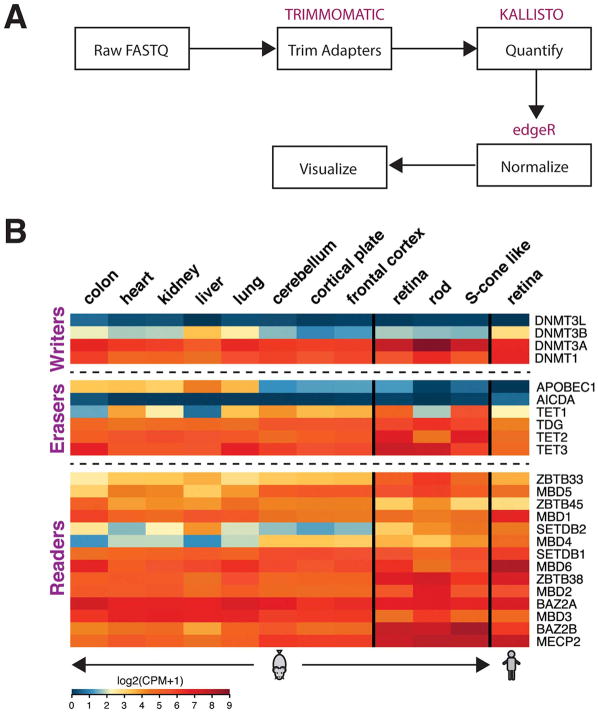
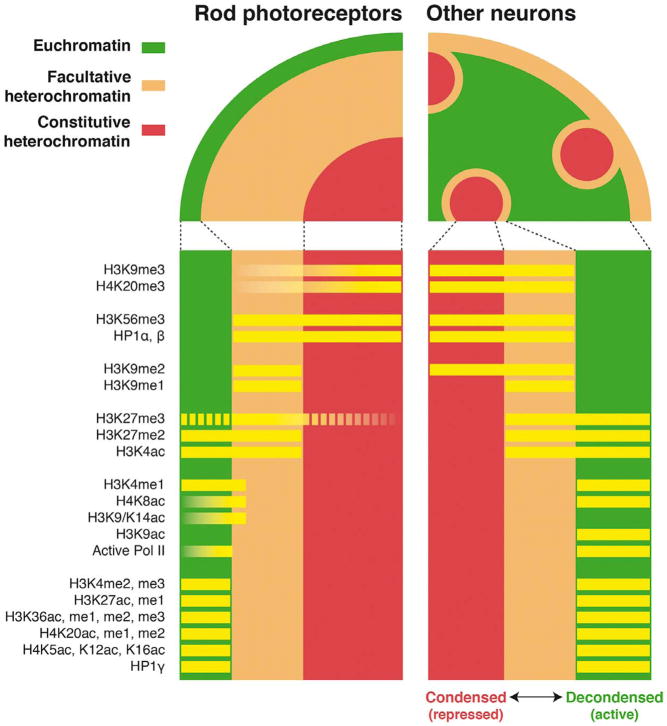
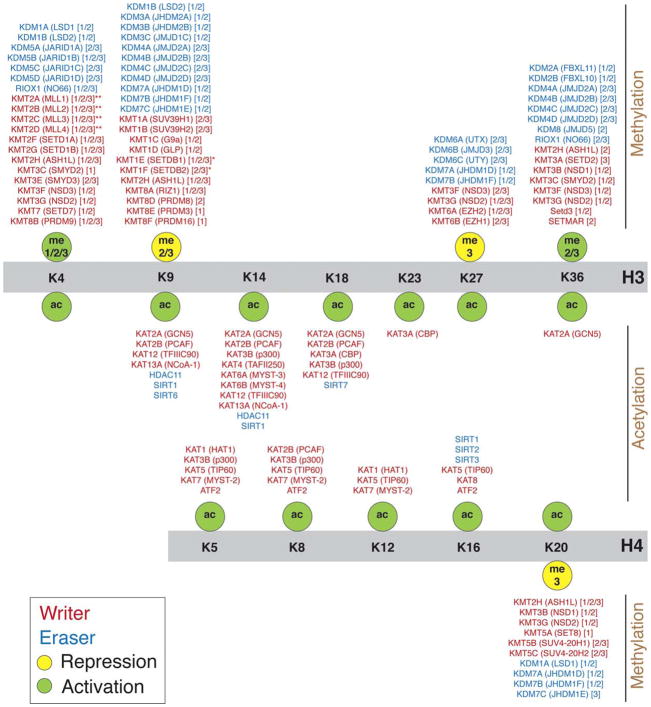
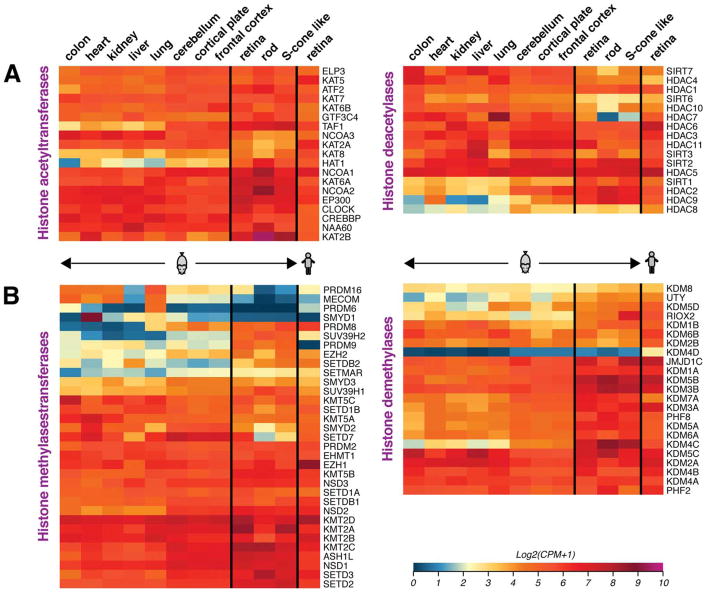
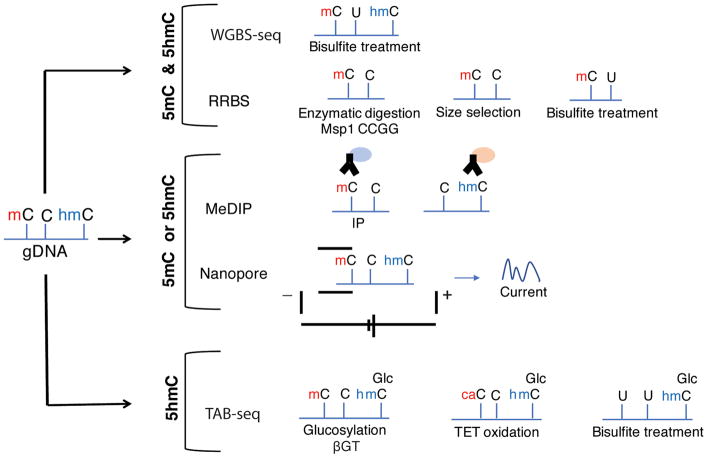
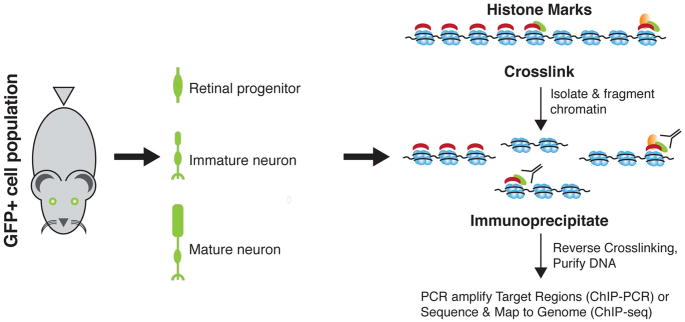
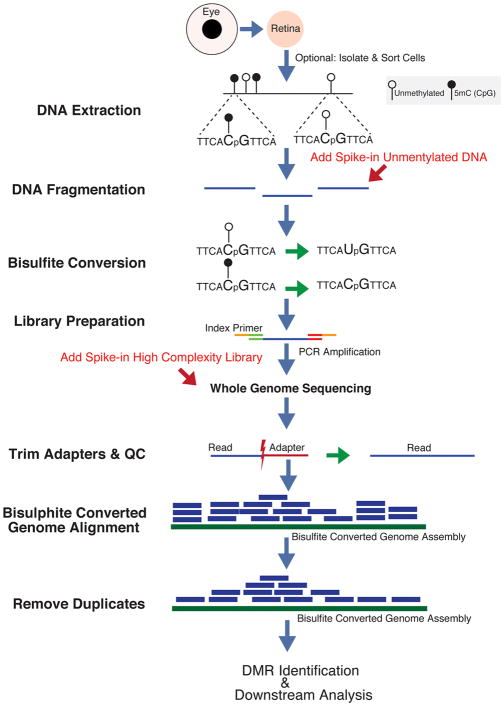
Complex biological processes, such as organogenesis and homeostasis, are stringently regulated by genetic programs that are fine-tuned by epigenetic factors to establish cell fates and/or to respond to the microenvironment. Gene regulatory networks that guide cell differentiation and function are modulated and stabilized by modifications to DNA, RNA and proteins. In this review, we focus on two key epigenetic changes - DNA methylation and histone modifications - and discuss their contribution to retinal development, aging and disease, especially in the context of age-related macular degeneration (AMD) and diabetic retinopathy. We highlight less-studied roles of DNA methylation and provide the RNA expression profiles of epigenetic enzymes in human and mouse retina in comparison to other tissues. We also review computational tools and emergent technologies to profile, analyze and integrate epigenetic information. We suggest implementation of editing tools and single-cell technologies to trace and perturb the epigenome for delineating its role in transcriptional regulation. Finally, we present our thoughts on exciting avenues for exploring epigenome in retinal metabolism, disease modeling, and regeneration.
Keywords: Chromatin; DNA methylation; Histone modification; Neuronal differentiation; Next generation sequencing; Photoreceptor; Retina neurodegeneration.
Published by Elsevier Ltd.
Figures
Similar articles
-
Epigenomics in stress tolerance of plants under the climate change.Mol Biol Rep. 2023 Jul;50(7):6201-6216. doi: 10.1007/s11033-023-08539-6. Epub 2023 Jun 9. Mol Biol Rep. 2023. PMID: 37294468 Review.
-
DNA methylation plays important roles in retinal development and diseases.Exp Eye Res. 2021 Oct;211:108733. doi: 10.1016/j.exer.2021.108733. Epub 2021 Aug 18. Exp Eye Res. 2021. PMID: 34418429 Review.
-
Epigenome-metabolism nexus in the retina: implications for aging and disease.Trends Genet. 2024 Aug;40(8):718-729. doi: 10.1016/j.tig.2024.04.012. Epub 2024 May 22. Trends Genet. 2024. PMID: 38782642 Free PMC article. Review.
-
QTL mapping of human retina DNA methylation identifies 87 gene-epigenome interactions in age-related macular degeneration.Nat Commun. 2024 Mar 4;15(1):1972. doi: 10.1038/s41467-024-46063-8. Nat Commun. 2024. PMID: 38438351 Free PMC article.
-
Potential of epigenetic mechanisms in AMD pathology.Front Biosci (Schol Ed). 2013 Jan 1;5(2):412-25. doi: 10.2741/s380. Front Biosci (Schol Ed). 2013. PMID: 23277059 Review.
Cited by
-
In utero Androgen Excess: A Developmental Commonality Preceding Polycystic Ovary Syndrome?Front Horm Res. 2019;53:1-17. doi: 10.1159/000494899. Epub 2019 Sep 9. Front Horm Res. 2019. PMID: 31499494 Free PMC article. Review.
-
Genome-wide DNA methylation analysis of extreme phenotypes in the identification of novel epigenetic modifications in diabetic retinopathy.Clin Epigenetics. 2022 Oct 31;14(1):137. doi: 10.1186/s13148-022-01354-z. Clin Epigenetics. 2022. PMID: 36316758 Free PMC article.
-
dnmt1 function is required to maintain retinal stem cells within the ciliary marginal zone of the zebrafish eye.Sci Rep. 2020 Jul 9;10(1):11293. doi: 10.1038/s41598-020-68016-z. Sci Rep. 2020. PMID: 32647199 Free PMC article.
-
Decrease of FZD4 exon 1 methylation in probands from FZD4-associated FEVR family of phenotypic heterogeneity.Front Med (Lausanne). 2022 Oct 24;9:976520. doi: 10.3389/fmed.2022.976520. eCollection 2022. Front Med (Lausanne). 2022. PMID: 36353221 Free PMC article.
-
Oxidative stress and epigenetics in ocular vascular aging: an updated review.Mol Med. 2023 Feb 27;29(1):28. doi: 10.1186/s10020-023-00624-7. Mol Med. 2023. PMID: 36849907 Free PMC article. Review.
References
-
- Agathocleous M, Harris WA. From progenitors to differentiated cells in the vertebrate retina. Annu Rev Cell Dev Biol. 2009;25:45–69. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
