

Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding
- PMID: 29545511
- PMCID: PMC7643870
- DOI: 10.1126/science.aam8999
Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding
Abstract
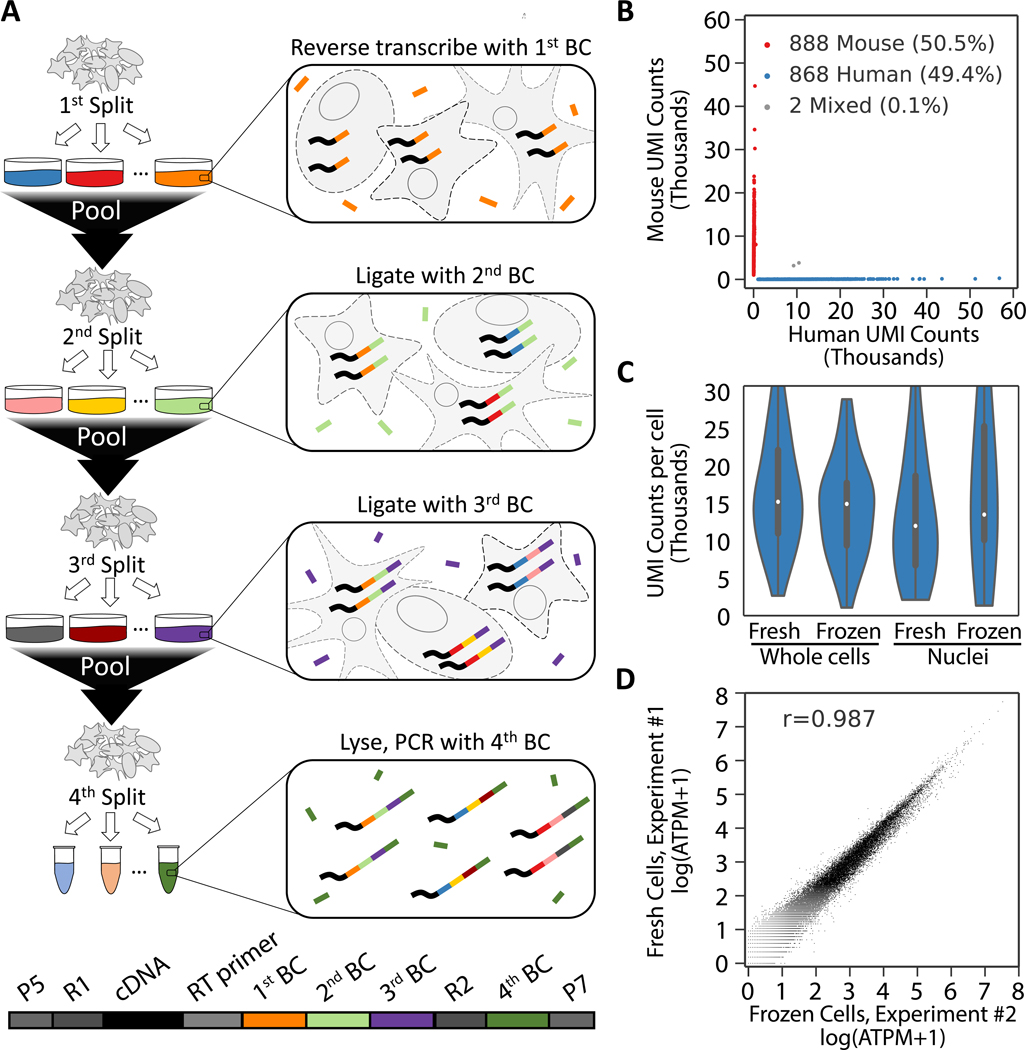
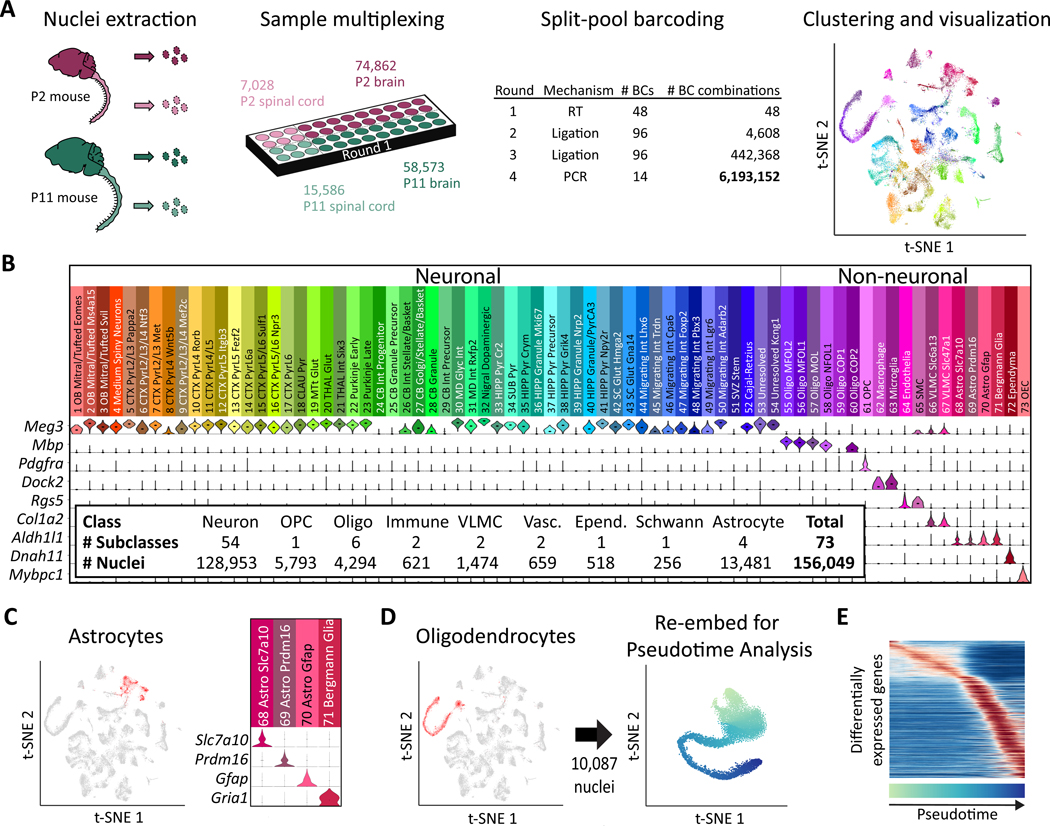
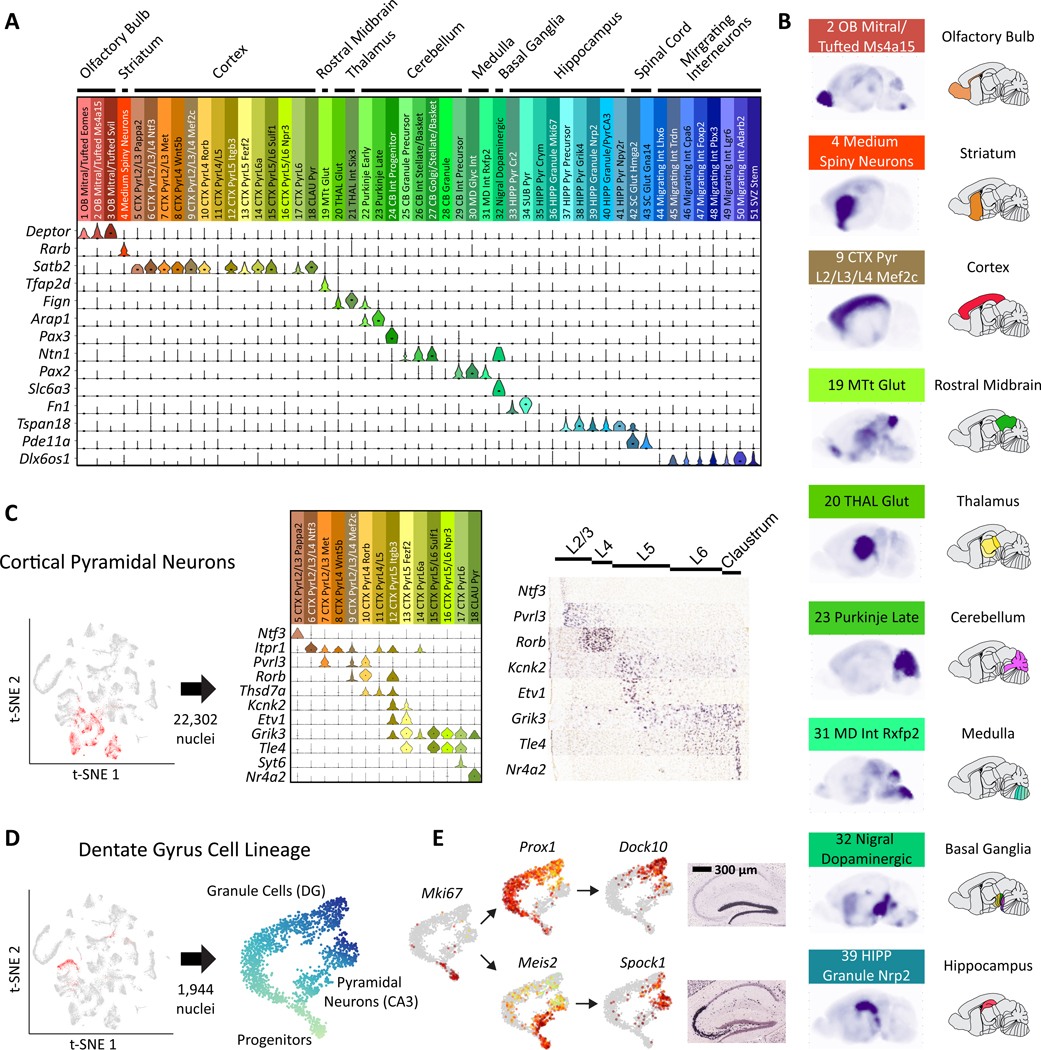
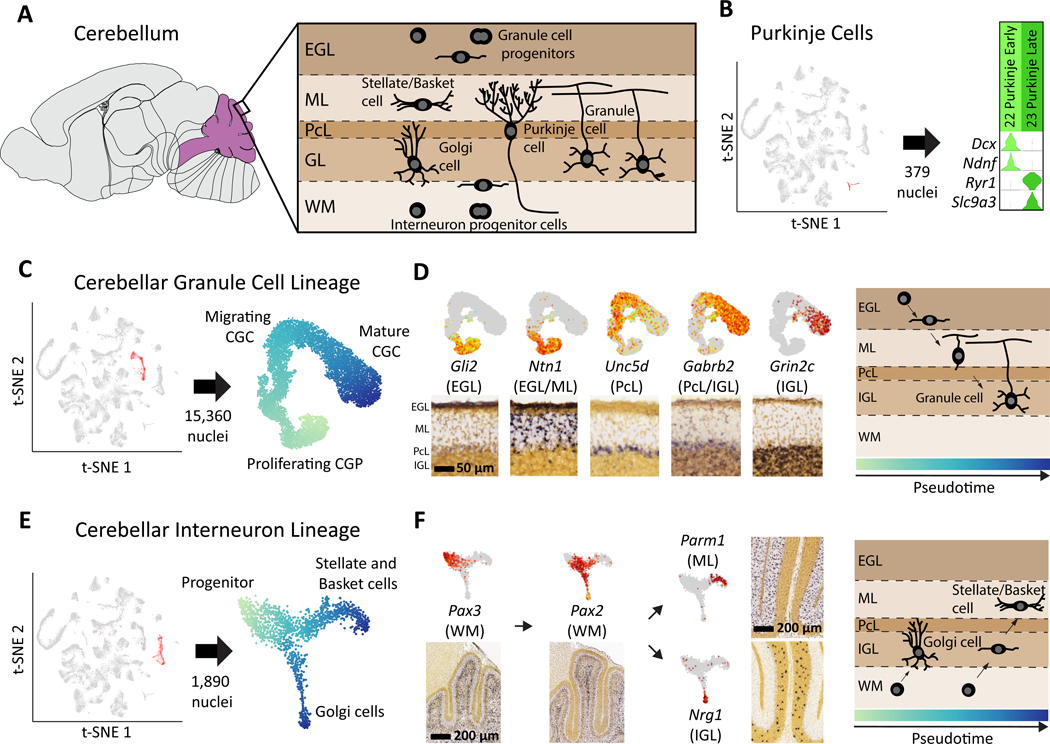
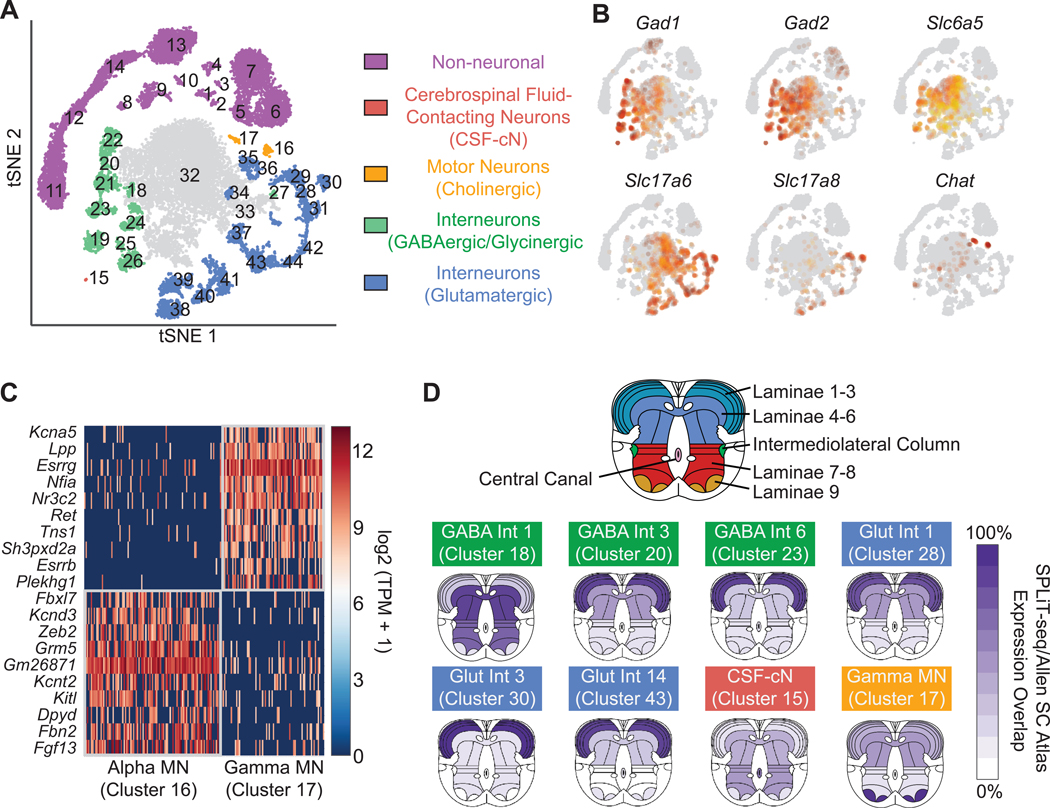
To facilitate scalable profiling of single cells, we developed split-pool ligation-based transcriptome sequencing (SPLiT-seq), a single-cell RNA-seq (scRNA-seq) method that labels the cellular origin of RNA through combinatorial barcoding. SPLiT-seq is compatible with fixed cells or nuclei, allows efficient sample multiplexing, and requires no customized equipment. We used SPLiT-seq to analyze 156,049 single-nucleus transcriptomes from postnatal day 2 and 11 mouse brains and spinal cords. More than 100 cell types were identified, with gene expression patterns corresponding to cellular function, regional specificity, and stage of differentiation. Pseudotime analysis revealed transcriptional programs driving four developmental lineages, providing a snapshot of early postnatal development in the murine central nervous system. SPLiT-seq provides a path toward comprehensive single-cell transcriptomic analysis of other similarly complex multicellular systems.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
