

TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia
- PMID: 29545601
- PMCID: PMC5854632
- DOI: 10.1038/s41598-018-22858-w
TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia
Abstract
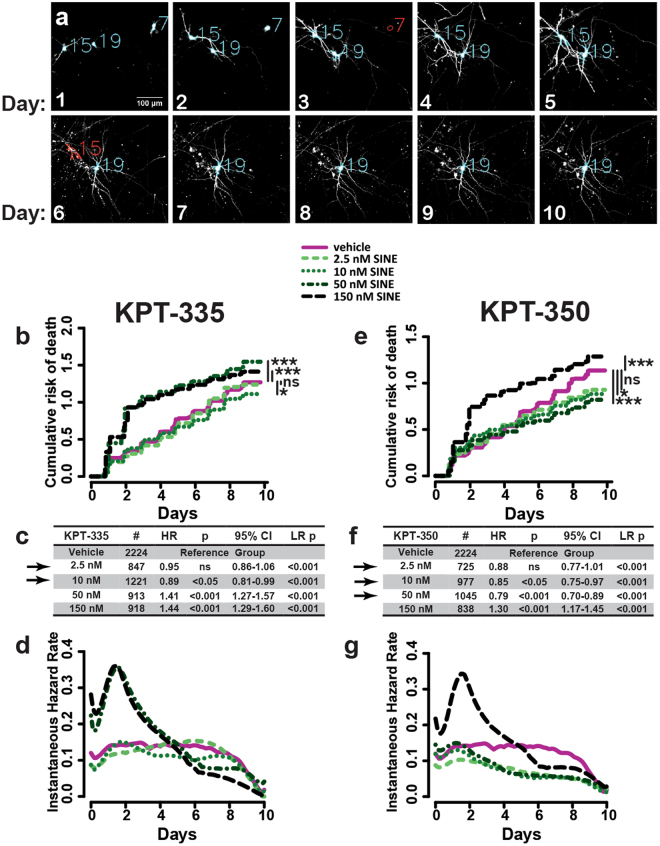
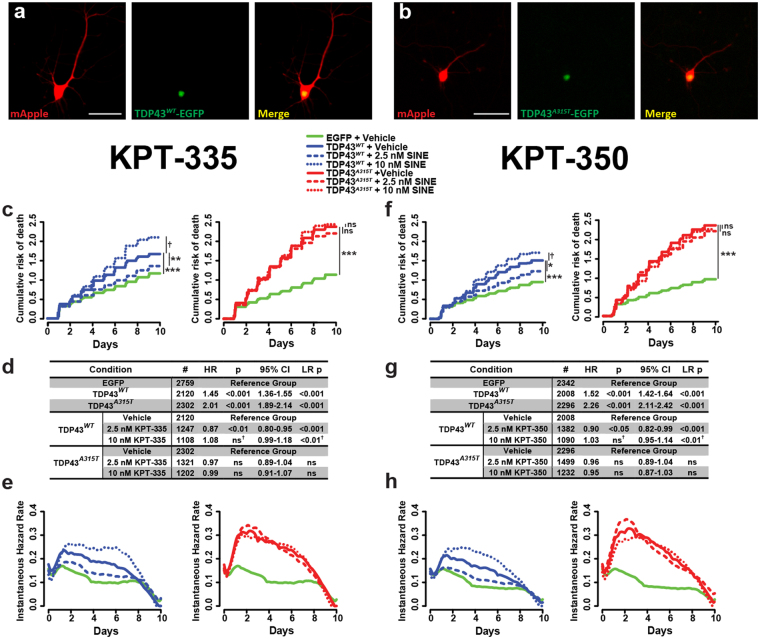
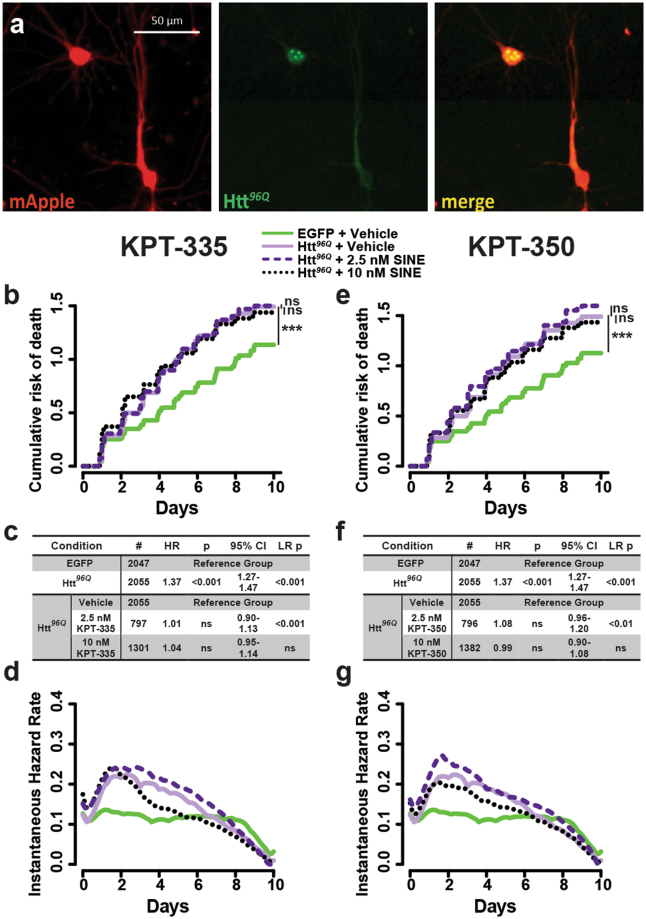
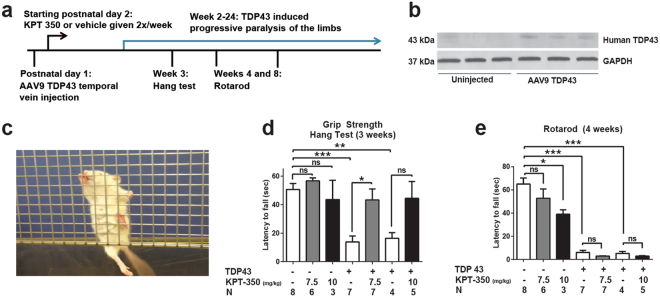
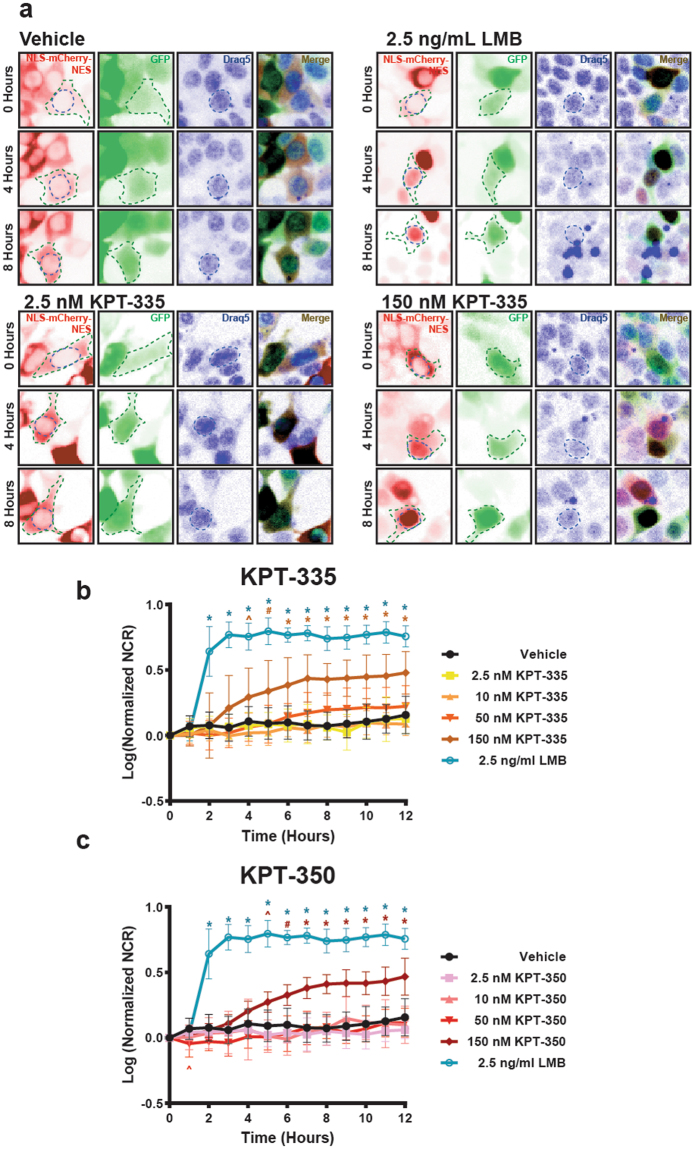
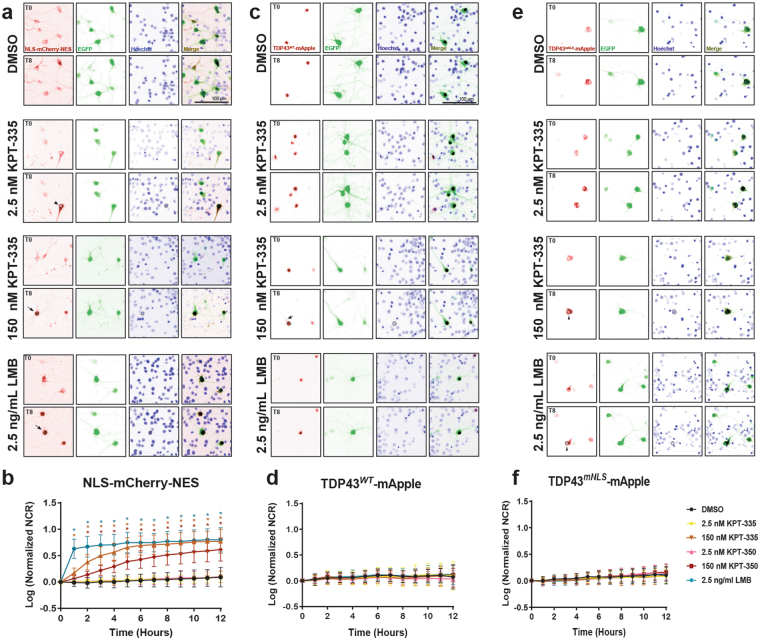
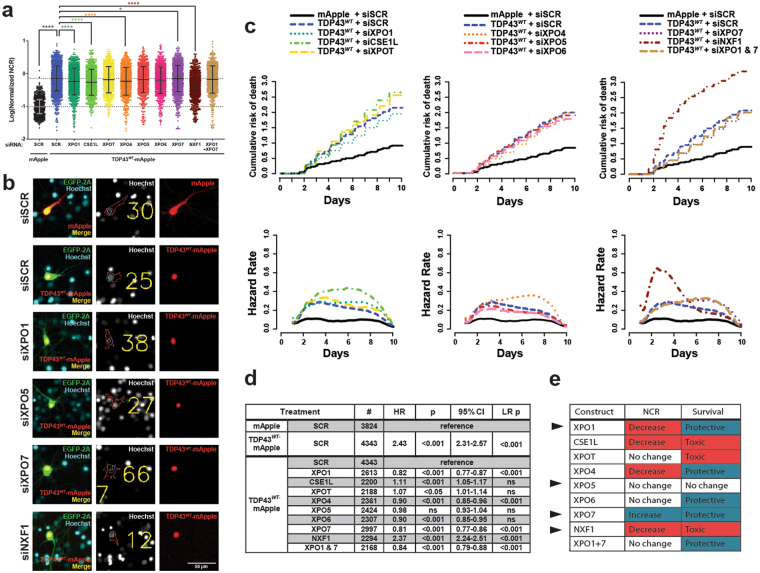
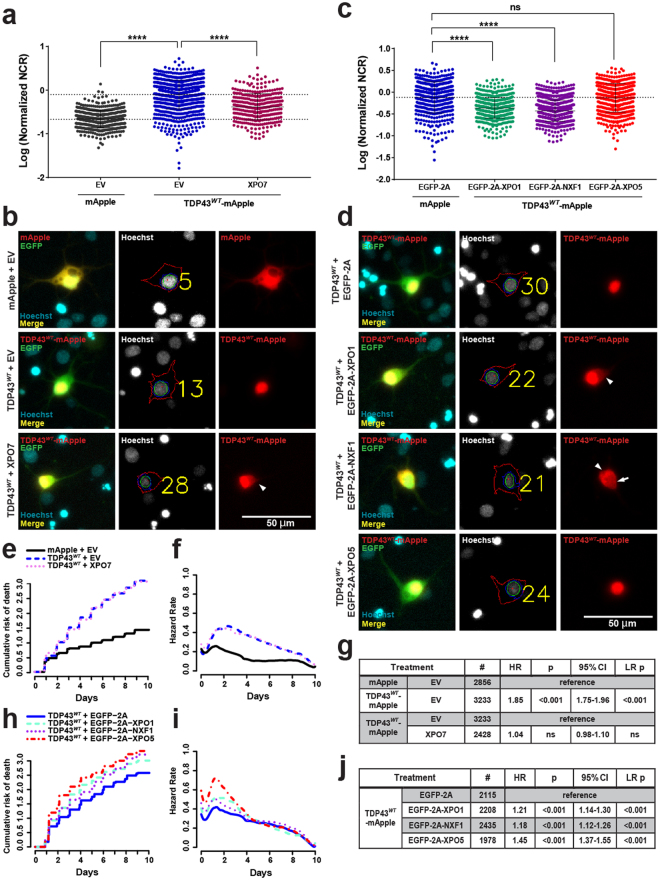
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are progressive neurodegenerative disorders marked in most cases by the nuclear exclusion and cytoplasmic deposition of the RNA binding protein TDP43. We previously demonstrated that ALS-associated mutant TDP43 accumulates within the cytoplasm, and that TDP43 mislocalization predicts neurodegeneration. Here, we sought to prevent neurodegeneration in ALS/FTD models using selective inhibitor of nuclear export (SINE) compounds that target exportin-1 (XPO1). SINE compounds modestly extend cellular survival in neuronal ALS/FTD models and mitigate motor symptoms in an in vivo rat ALS model. At high doses, SINE compounds block nuclear egress of an XPO1 cargo reporter, but not at lower concentrations that were associated with neuroprotection. Neither SINE compounds nor leptomycin B, a separate XPO1 inhibitor, enhanced nuclear TDP43 levels, while depletion of XPO1 or other exportins had little effect on TDP43 localization, suggesting that no single exporter is necessary for TDP43 export. Supporting this hypothesis, we find overexpression of XPO1, XPO7 and NXF1 are each sufficient to promote nuclear TDP43 egress. Taken together, our results indicate that redundant pathways regulate TDP43 nuclear export, and that therapeutic prevention of cytoplasmic TDP43 accumulation in ALS/FTD may be enhanced by targeting several overlapping mechanisms.
Conflict of interest statement
S.T. is an employee of Karyopharm Therapeutics, and has financial interest in this company. The remaining authors have no competing interests to declare.
Figures
References
-
- Charcot JM, Joffory A. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arch. Physiol. Neurol. Pathol. 1869;2:744–754.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
