

VDAC Regulation: A Mitochondrial Target to Stop Cell Proliferation
- PMID: 29551129
- PMCID: PMC6234976
- DOI: 10.1016/bs.acr.2018.02.002
VDAC Regulation: A Mitochondrial Target to Stop Cell Proliferation
Abstract
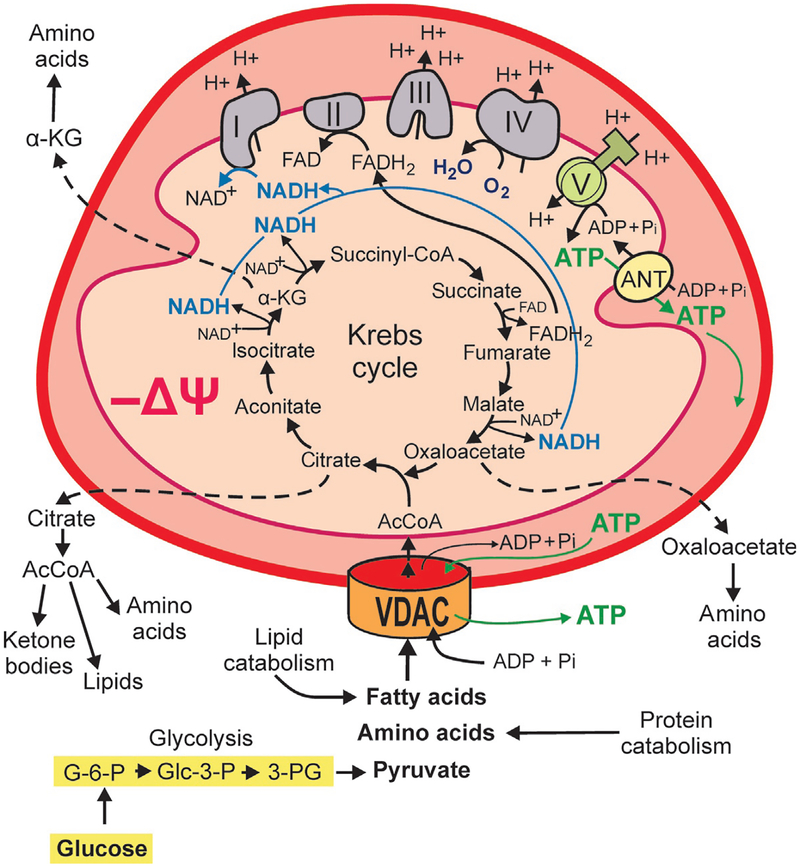
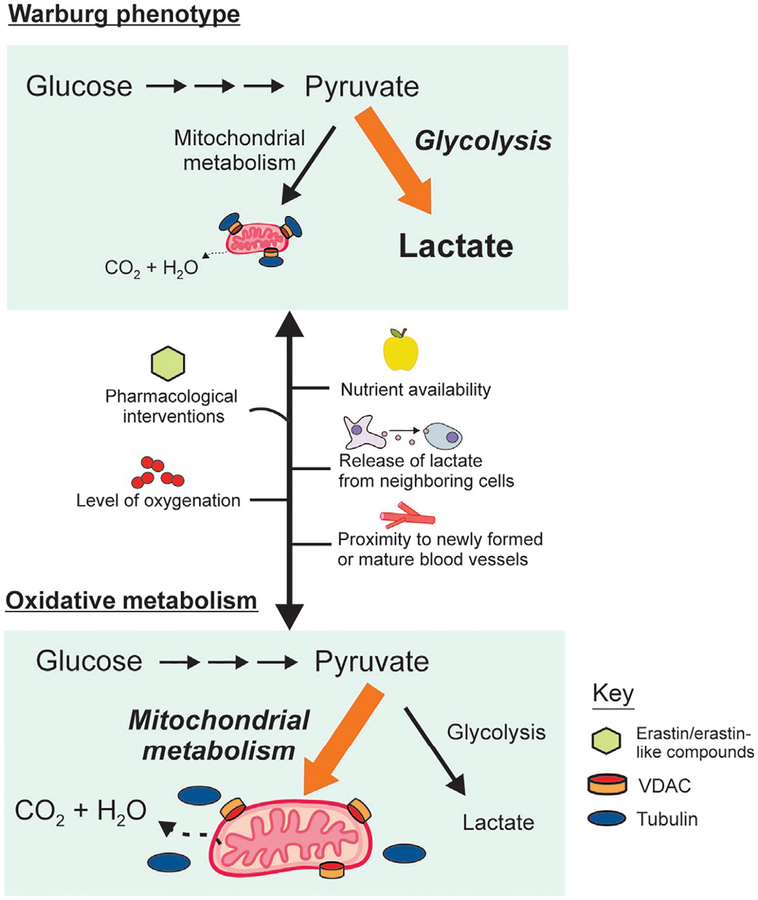
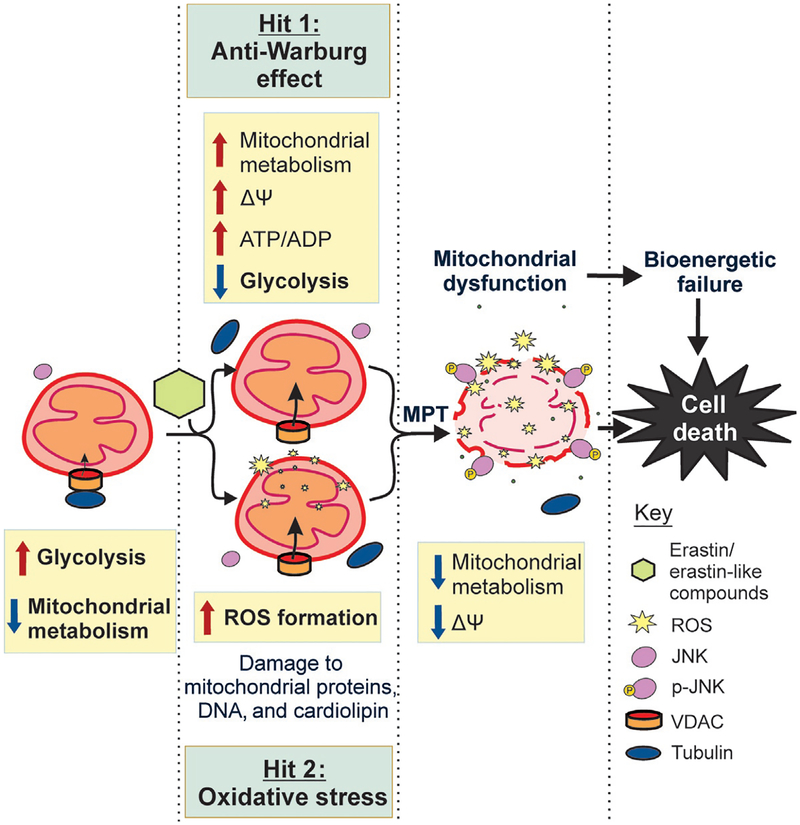
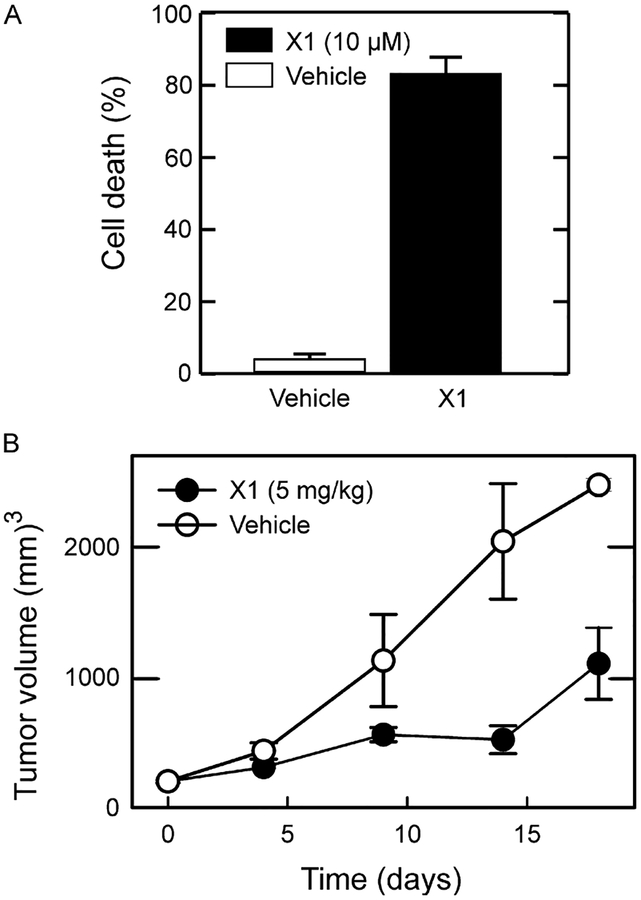
Cancer metabolism is emerging as a chemotherapeutic target. Enhanced glycolysis and suppression of mitochondrial metabolism characterize the Warburg phenotype in cancer cells. The flux of respiratory substrates, ADP, and Pi into mitochondria and the release of mitochondrial ATP to the cytosol occur through voltage-dependent anion channels (VDACs) located in the mitochondrial outer membrane. Catabolism of respiratory substrates in the Krebs cycle generates NADH and FADH2 that enter the electron transport chain (ETC) to generate a proton motive force that maintains mitochondrial membrane potential (ΔΨ) and is utilized to generate ATP. The ETC is also the major cellular source of mitochondrial reactive oxygen species (ROS). αβ-Tubulin heterodimers decrease VDAC conductance in lipid bilayers. High constitutive levels of cytosolic free tubulin in intact cancer cells close VDAC decreasing mitochondrial ΔΨ and mitochondrial metabolism. The VDAC-tubulin interaction regulates VDAC opening and globally controls mitochondrial metabolism, ROS formation, and the intracellular flow of energy. Erastin, a VDAC-binding molecule lethal to some cancer cell types, and erastin-like compounds identified in a high-throughput screening antagonize the inhibitory effect of tubulin on VDAC. Reversal of tubulin inhibition of VDAC increases VDAC conductance and the flux of metabolites into and out of mitochondria. VDAC opening promotes a higher mitochondrial ΔΨ and a global increase in mitochondrial metabolism leading to high cytosolic ATP/ADP ratios that inhibit glycolysis. VDAC opening also increases ROS production causing oxidative stress that, in turn, leads to mitochondrial dysfunction, bioenergetic failure, and cell death. In summary, antagonism of the VDAC-tubulin interaction promotes cell death by a "double-hit model" characterized by reversion of the proproliferative Warburg phenotype (anti-Warburg) and promotion of oxidative stress.
Keywords: Cancer metabolism; Erastin; Glycolysis; Mitochondria; Oxidative stress; Tubulin; Voltage-dependent anion channel; Warburg effect.
© 2018 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
VDAC-Tubulin, an Anti-Warburg Pro-Oxidant Switch.Front Oncol. 2017 Jan 23;7:4. doi: 10.3389/fonc.2017.00004. eCollection 2017. Front Oncol. 2017. PMID: 28168164 Free PMC article. Review.
-
Erastin-Like Anti-Warburg Agents Prevent Mitochondrial Depolarization Induced by Free Tubulin and Decrease Lactate Formation in Cancer Cells.SLAS Discov. 2018 Jan;23(1):23-33. doi: 10.1177/2472555217731556. Epub 2017 Oct 12. SLAS Discov. 2018. PMID: 29024608 Free PMC article.
-
Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells.Biochem Pharmacol. 2018 Feb;148:155-162. doi: 10.1016/j.bcp.2017.12.022. Epub 2017 Dec 28. Biochem Pharmacol. 2018. PMID: 29289511 Free PMC article.
-
Warburg revisited: regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells.J Pharmacol Exp Ther. 2012 Sep;342(3):637-41. doi: 10.1124/jpet.112.192153. Epub 2012 Jun 13. J Pharmacol Exp Ther. 2012. PMID: 22700429 Free PMC article. Review.
-
ATP/ADP ratio, the missed connection between mitochondria and the Warburg effect.Mitochondrion. 2014 Nov;19 Pt A:78-84. doi: 10.1016/j.mito.2014.09.002. Epub 2014 Sep 16. Mitochondrion. 2014. PMID: 25229666 Free PMC article. Review.
Cited by
-
Inflammatory IFIT3 renders chemotherapy resistance by regulating post-translational modification of VDAC2 in pancreatic cancer.Theranostics. 2020 Jun 1;10(16):7178-7192. doi: 10.7150/thno.43093. eCollection 2020. Theranostics. 2020. PMID: 32641986 Free PMC article.
-
Melatonin regulates mitochondrial function to alleviate ferroptosis through the MT2/Akt signaling pathway in swine testicular cells.Sci Rep. 2024 Jul 2;14(1):15215. doi: 10.1038/s41598-024-65666-1. Sci Rep. 2024. PMID: 38956409 Free PMC article.
-
[Gefitinib inhibits glycolysis and induces programmed cell death in non-small cell lung cancer cells].Nan Fang Yi Ke Da Xue Xue Bao. 2020 Jun 30;40(6):884-892. doi: 10.12122/j.issn.1673-4254.2020.06.17. Nan Fang Yi Ke Da Xue Xue Bao. 2020. PMID: 32895203 Free PMC article. Chinese.
-
Ferroptosis: Mechanism and connections with cutaneous diseases.Front Cell Dev Biol. 2023 Jan 4;10:1079548. doi: 10.3389/fcell.2022.1079548. eCollection 2022. Front Cell Dev Biol. 2023. PMID: 36684424 Free PMC article. Review.
-
Protective Effect of Treated Olive Mill Wastewater on Target Bacteria and Mitochondrial Voltage-Dependent Anion-Selective Channel 1.Antioxidants (Basel). 2023 Jan 30;12(2):322. doi: 10.3390/antiox12020322. Antioxidants (Basel). 2023. PMID: 36829881 Free PMC article.
References
-
- Al Jamal JA (2005). Involvement of porin N,N-dicyclohexylcarbodiimide-reactive domain in hexokinase binding to the outer mitochondrial membrane. The Protein Journal, 24, 1–8. - PubMed
-
- Beckner ME, Gobbel GT, Abounader R, Burovic F, Agostino NR, Laterra J, et al. (2005). Glycolytic glioma cells with active glycogen synthase are sensitive to PTEN and inhibitors of PI3K and gluconeogenesis. Laboratory Investigation, 85, 1457–1470. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
