

Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection
- PMID: 29553575
- PMCID: PMC6118326
- DOI: 10.1038/nbt.4110
Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection
Abstract
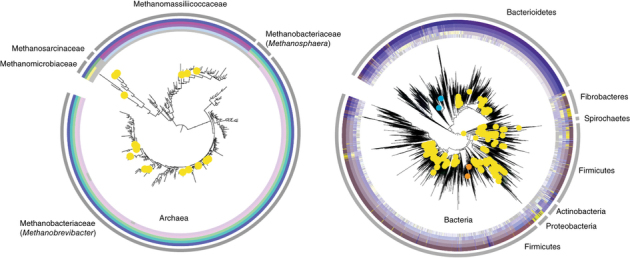
Productivity of ruminant livestock depends on the rumen microbiota, which ferment indigestible plant polysaccharides into nutrients used for growth. Understanding the functions carried out by the rumen microbiota is important for reducing greenhouse gas production by ruminants and for developing biofuels from lignocellulose. We present 410 cultured bacteria and archaea, together with their reference genomes, representing every cultivated rumen-associated archaeal and bacterial family. We evaluate polysaccharide degradation, short-chain fatty acid production and methanogenesis pathways, and assign specific taxa to functions. A total of 336 organisms were present in available rumen metagenomic data sets, and 134 were present in human gut microbiome data sets. Comparison with the human microbiome revealed rumen-specific enrichment for genes encoding de novo synthesis of vitamin B12, ongoing evolution by gene loss and potential vertical inheritance of the rumen microbiome based on underrepresentation of markers of environmental stress. We estimate that our Hungate genome resource represents ∼75% of the genus-level bacterial and archaeal taxa present in the rumen.
Conflict of interest statement
The authors declare no competing financial interests.
Figures




References
-
- Godfray HC, et al. Food security: the challenge of feeding 9 billion people. Science. 2010;327:812–818. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
