

Autophagy during viral infection - a double-edged sword
- PMID: 29556036
- PMCID: PMC6907743
- DOI: 10.1038/s41579-018-0003-6
Autophagy during viral infection - a double-edged sword
Abstract
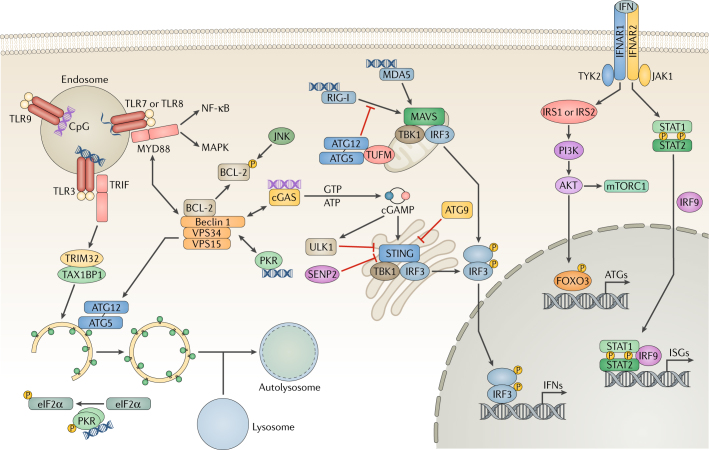
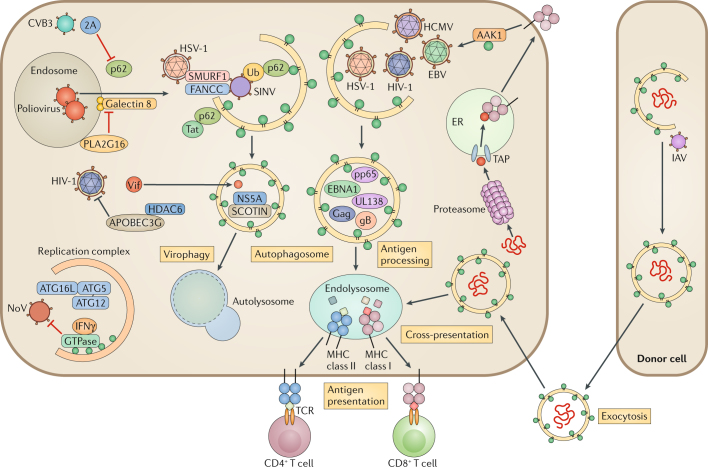
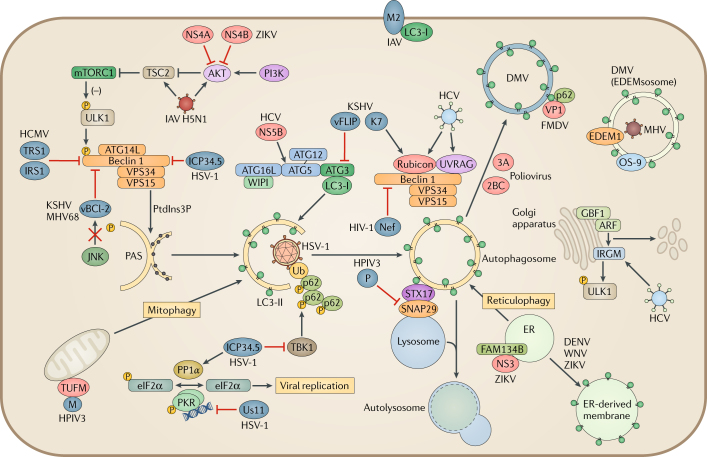
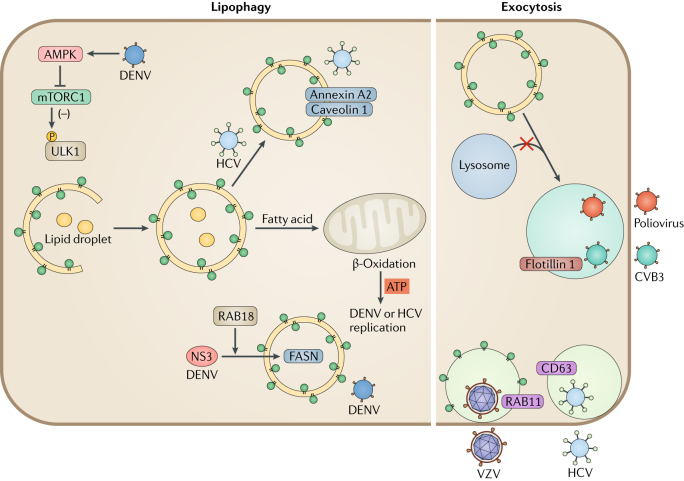
Autophagy is a powerful tool that host cells use to defend against viral infection. Double-membrane vesicles, termed autophagosomes, deliver trapped viral cargo to the lysosome for degradation. Specifically, autophagy initiates an innate immune response by cooperating with pattern recognition receptor signalling to induce interferon production. It also selectively degrades immune components associated with viral particles. Following degradation, autophagy coordinates adaptive immunity by delivering virus-derived antigens for presentation to T lymphocytes. However, in an ongoing evolutionary arms race, viruses have acquired the potent ability to hijack and subvert autophagy for their benefit. In this Review, we focus on the key regulatory steps during viral infection in which autophagy is involved and discuss the specific molecular mechanisms that diverse viruses use to repurpose autophagy for their life cycle and pathogenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
