

FusorSV: an algorithm for optimally combining data from multiple structural variation detection methods
- PMID: 29559002
- PMCID: PMC5859555
- DOI: 10.1186/s13059-018-1404-6
FusorSV: an algorithm for optimally combining data from multiple structural variation detection methods
Abstract
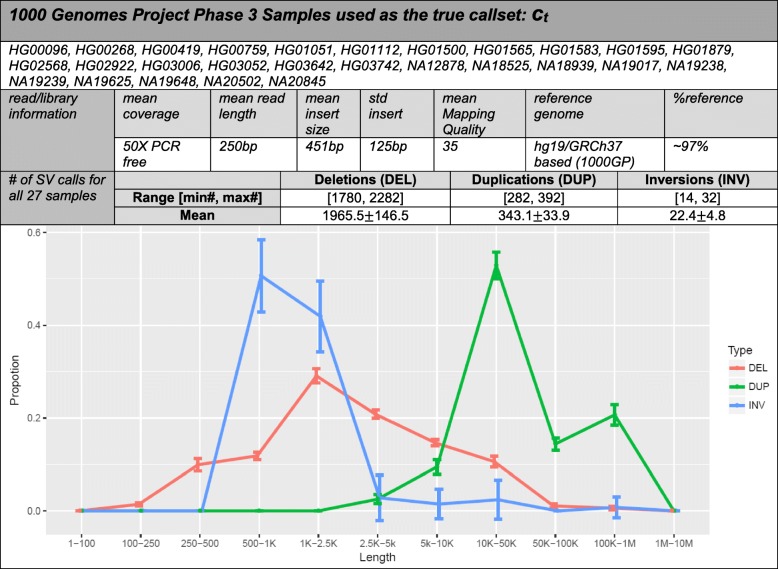
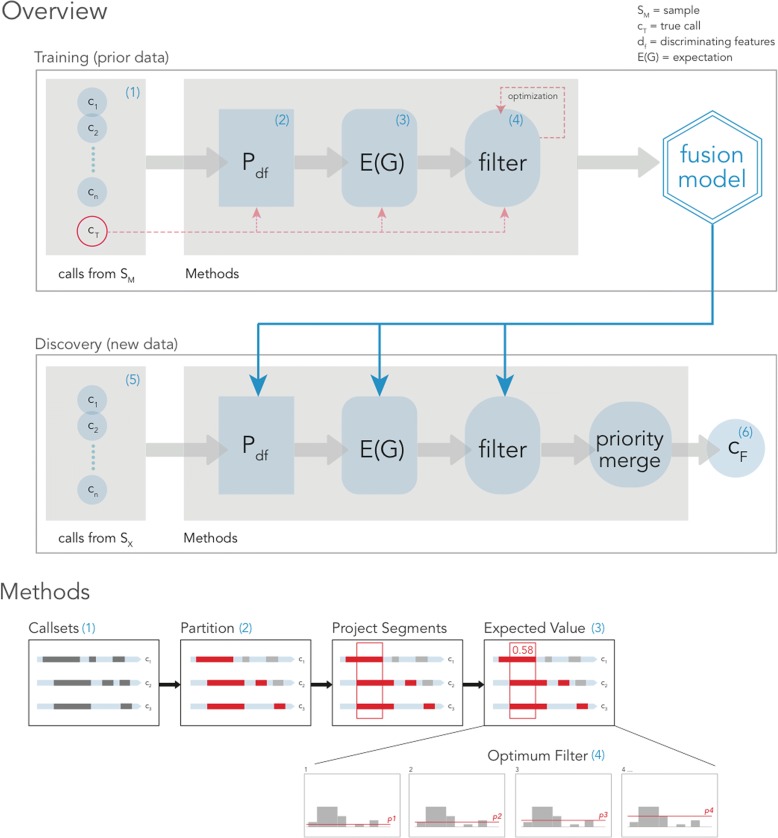
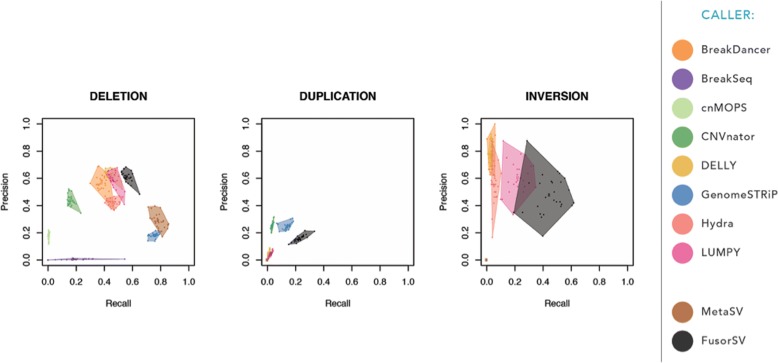
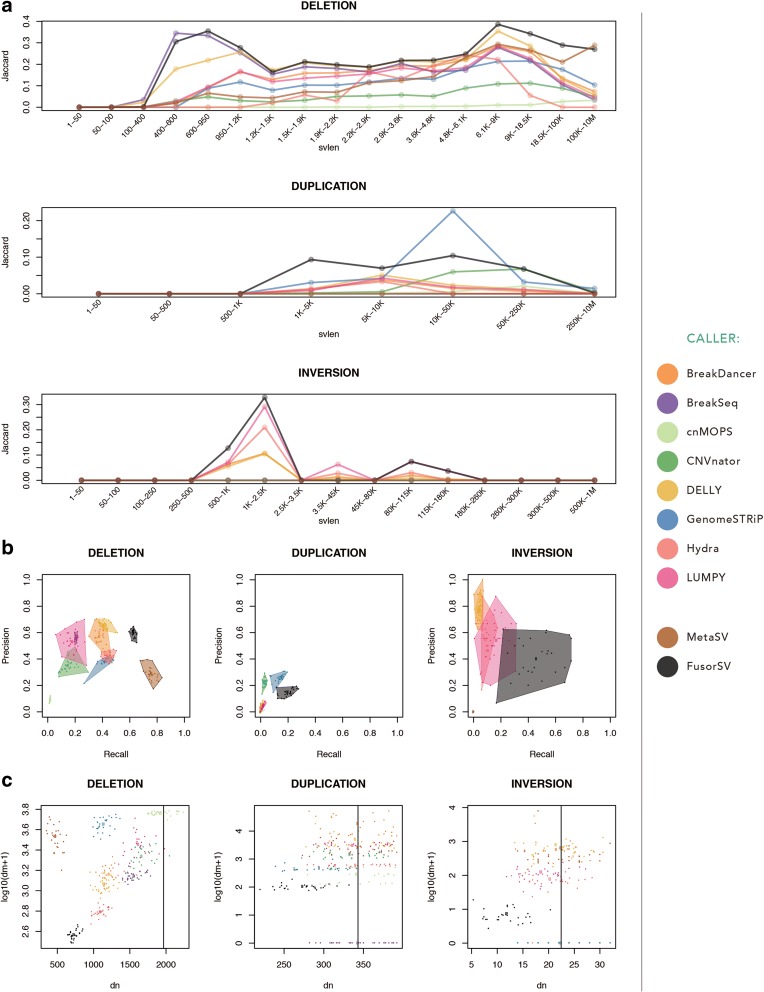
Comprehensive and accurate identification of structural variations (SVs) from next generation sequencing data remains a major challenge. We develop FusorSV, which uses a data mining approach to assess performance and merge callsets from an ensemble of SV-calling algorithms. It includes a fusion model built using analysis of 27 deep-coverage human genomes from the 1000 Genomes Project. We identify 843 novel SV calls that were not reported by the 1000 Genomes Project for these 27 samples. Experimental validation of a subset of these calls yields a validation rate of 86.7%. FusorSV is available at https://github.com/TheJacksonLaboratory/SVE .
Keywords: Copy number variation; Genome rearrangements; Next generation sequencing; Structural variation.
Conflict of interest statement
Ethics approval and consent to participate
The data from the 27 germline genomes used in this project have been consented and approved for further analysis as part of the 1000 Genomes Project.
Consent for publication
The data from the 27 germline genomes used in this project have been consented and approved for publication as part of the 1000 Genomes Project.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
