

Chromium disrupts chromatin organization and CTCF access to its cognate sites in promoters of differentially expressed genes
- PMID: 29561703
- PMCID: PMC6140807
- DOI: 10.1080/15592294.2018.1454243
Chromium disrupts chromatin organization and CTCF access to its cognate sites in promoters of differentially expressed genes
Abstract
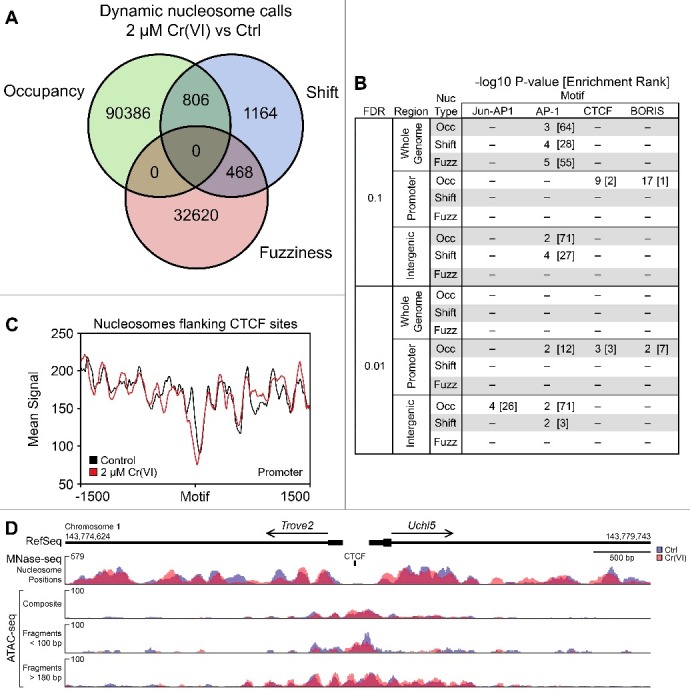
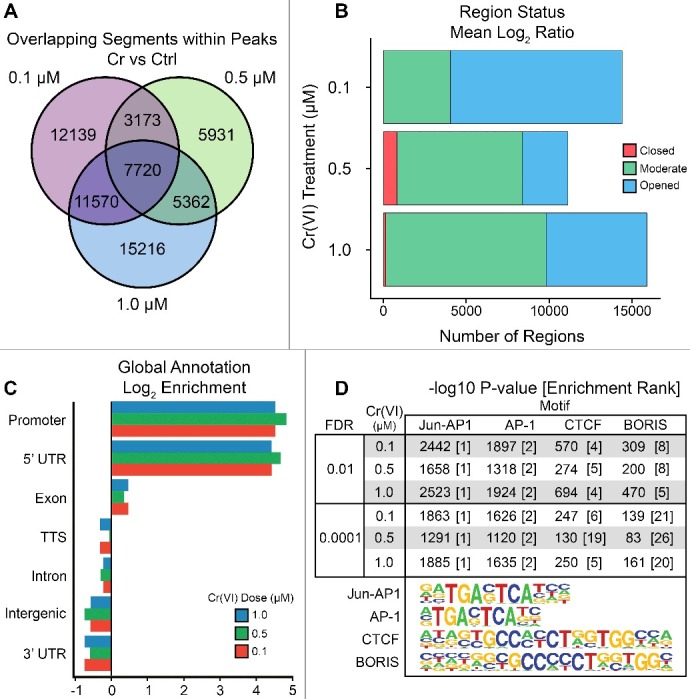
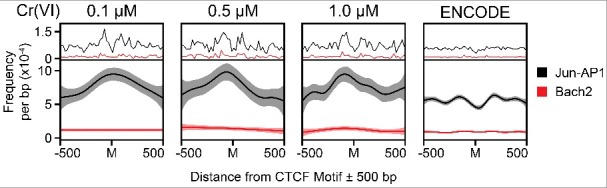
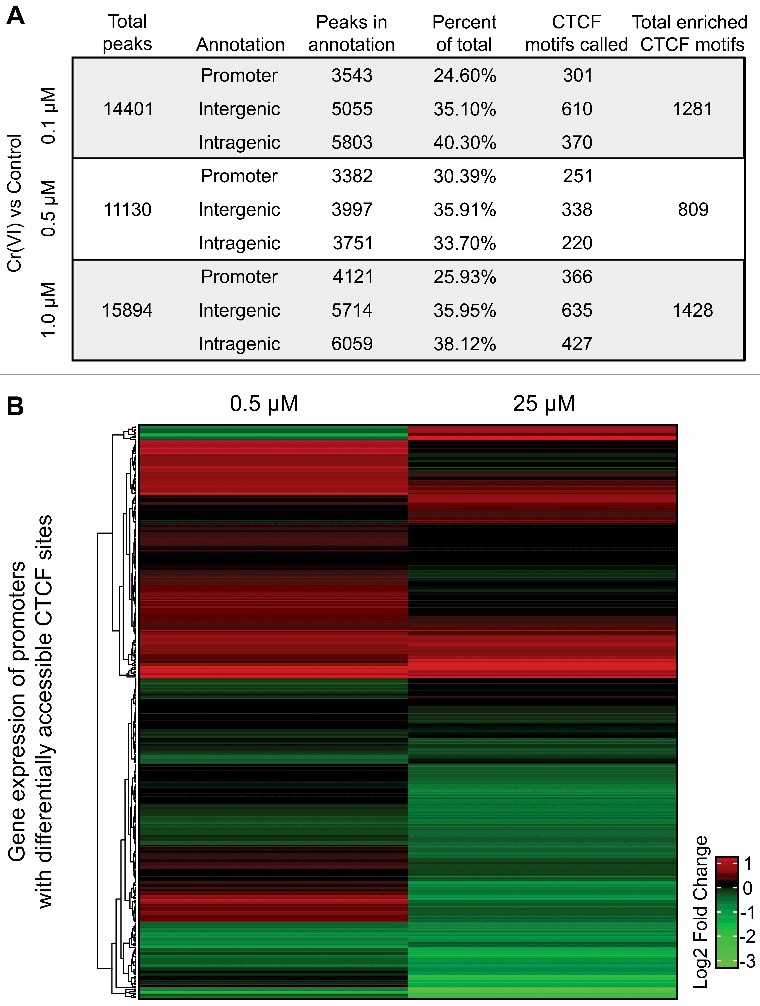
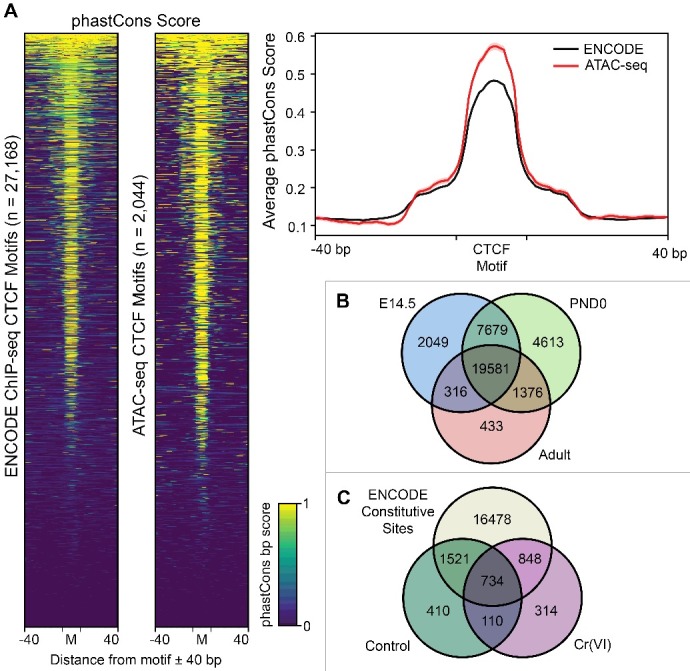
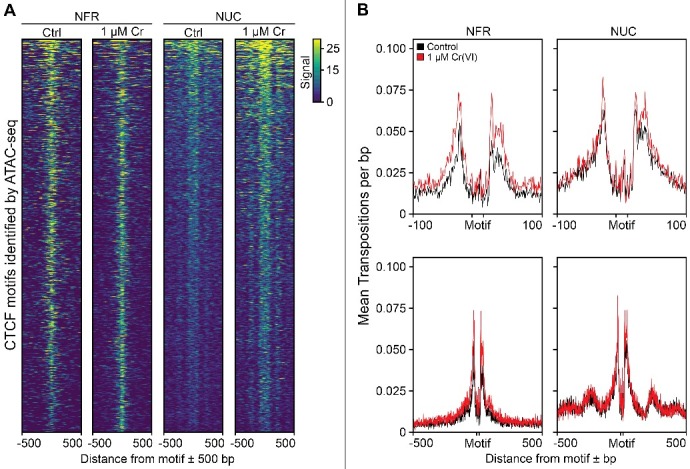
Hexavalent chromium compounds are well-established respiratory carcinogens used in industrial processes. While inhalation exposure constitutes an occupational risk affecting mostly chromium workers, environmental exposure from drinking water is a widespread gastrointestinal cancer risk, affecting millions of people throughout the world. Cr(VI) is genotoxic, forming protein-Cr-DNA adducts and silencing tumor suppressor genes, but its mechanism of action at the molecular level is poorly understood. Our prior work using FAIRE showed that Cr(VI) disrupted the binding of transcription factors CTCF and AP-1 to their cognate chromatin sites. Here, we used two complementary approaches to test the hypothesis that chromium perturbs chromatin organization and dynamics. DANPOS2 analyses of MNase-seq data identified several chromatin alterations induced by Cr(VI) affecting nucleosome architecture, including occupancy changes at specific genome locations; position shifts of 10 nucleotides or more; and changes in position amplitude or fuzziness. ATAC-seq analysis revealed that Cr(VI) disrupted the accessibility of chromatin regions enriched for CTCF and AP-1 binding motifs, with a significant co-occurrence of binding sites for both factors in the same region. Cr(VI)-enriched CTCF sites were confirmed by ChIP-seq and found to correlate with evolutionarily conserved sites occupied by CTCF in vivo, as determined by comparison with ENCODE-validated CTCF datasets from mouse liver. In addition, more than 30% of the Cr(VI)-enriched CTCF sites were located in promoters of genes differentially expressed from chromium treatment. Our results support the conclusion that Cr(VI) exposure promotes broad changes in chromatin accessibility and suggest that the subsequent effects on transcription regulation may result from disruption of CTCF binding and nucleosome spacing, implicating transcription regulatory mechanisms as primary Cr(VI) targets.
Keywords: ATAC-seq; CTCF; chromatin organization; hexavalent chromium; nucleosome architecture; transcription.
Figures
Similar articles
-
Hexavalent chromium promotes differential binding of CTCF to its cognate sites in Euchromatin.Epigenetics. 2021 Dec;16(12):1361-1376. doi: 10.1080/15592294.2020.1864168. Epub 2021 Jan 7. Epigenetics. 2021. PMID: 33319643 Free PMC article.
-
Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE) analysis uncovers broad changes in chromatin structure resulting from hexavalent chromium exposure.PLoS One. 2014 May 16;9(5):e97849. doi: 10.1371/journal.pone.0097849. eCollection 2014. PLoS One. 2014. PMID: 24837440 Free PMC article.
-
The Dynamic Chromatin Architecture of the Regenerating Liver.Cell Mol Gastroenterol Hepatol. 2020;9(1):121-143. doi: 10.1016/j.jcmgh.2019.09.006. Epub 2019 Oct 17. Cell Mol Gastroenterol Hepatol. 2020. PMID: 31629814 Free PMC article.
-
Hexavalent chromium disrupts chromatin architecture.Semin Cancer Biol. 2021 Nov;76:54-60. doi: 10.1016/j.semcancer.2021.07.009. Epub 2021 Jul 15. Semin Cancer Biol. 2021. PMID: 34274487 Free PMC article. Review.
-
Discovering a binary CTCF code with a little help from BORIS.Nucleus. 2018 Jan 1;9(1):33-41. doi: 10.1080/19491034.2017.1394536. Epub 2017 Dec 5. Nucleus. 2018. PMID: 29077515 Free PMC article. Review.
Cited by
-
The Effect of Hexavalent Chromium on the Incidence and Mortality of Human Cancers: A Meta-Analysis Based on Published Epidemiological Cohort Studies.Front Oncol. 2019 Feb 4;9:24. doi: 10.3389/fonc.2019.00024. eCollection 2019. Front Oncol. 2019. PMID: 30778374 Free PMC article.
-
Alcohol induced increases in sperm Histone H3 lysine 4 trimethylation correlate with increased placental CTCF occupancy and altered developmental programming.Sci Rep. 2022 May 25;12(1):8839. doi: 10.1038/s41598-022-12188-3. Sci Rep. 2022. PMID: 35614060 Free PMC article.
-
Hexavalent Chromium Targets Securin to Drive Numerical Chromosome Instability in Human Lung Cells.Int J Mol Sci. 2023 Dec 23;25(1):256. doi: 10.3390/ijms25010256. Int J Mol Sci. 2023. PMID: 38203427 Free PMC article.
-
Chromium as a Risk Factor for Breast Cancer: A Meta-Analysis.Asian Pac J Cancer Prev. 2022 Dec 1;23(12):3993-4003. doi: 10.31557/APJCP.2022.23.12.3993. Asian Pac J Cancer Prev. 2022. PMID: 36579979 Free PMC article. Review.
-
Chromium exposure disrupts chromatin architecture upsetting the mechanisms that regulate transcription.Exp Biol Med (Maywood). 2019 Jun;244(9):752-757. doi: 10.1177/1535370219839953. Epub 2019 Apr 1. Exp Biol Med (Maywood). 2019. PMID: 30935235 Free PMC article. Review.
References
-
- Wilbur S, Abadin H, Fay M, et al. Toxicological profile for chromium. Atlanta: (GA: ): ATSDR, CDC; 2012. - PubMed
-
- National Toxicology P Toxicology and carcinogenesis studies of sodium dichromate dihydrate (Cas No. 7789-12-0) in F344/N rats and B6C3F1 mice (drinking water studies). Natl Toxicol Program Tech Rep Ser. 2008:1–192. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources