De novo mutations in regulatory elements in neurodevelopmental disorders
- PMID: 29562236
- PMCID: PMC5912909
- DOI: 10.1038/nature25983
De novo mutations in regulatory elements in neurodevelopmental disorders
Abstract
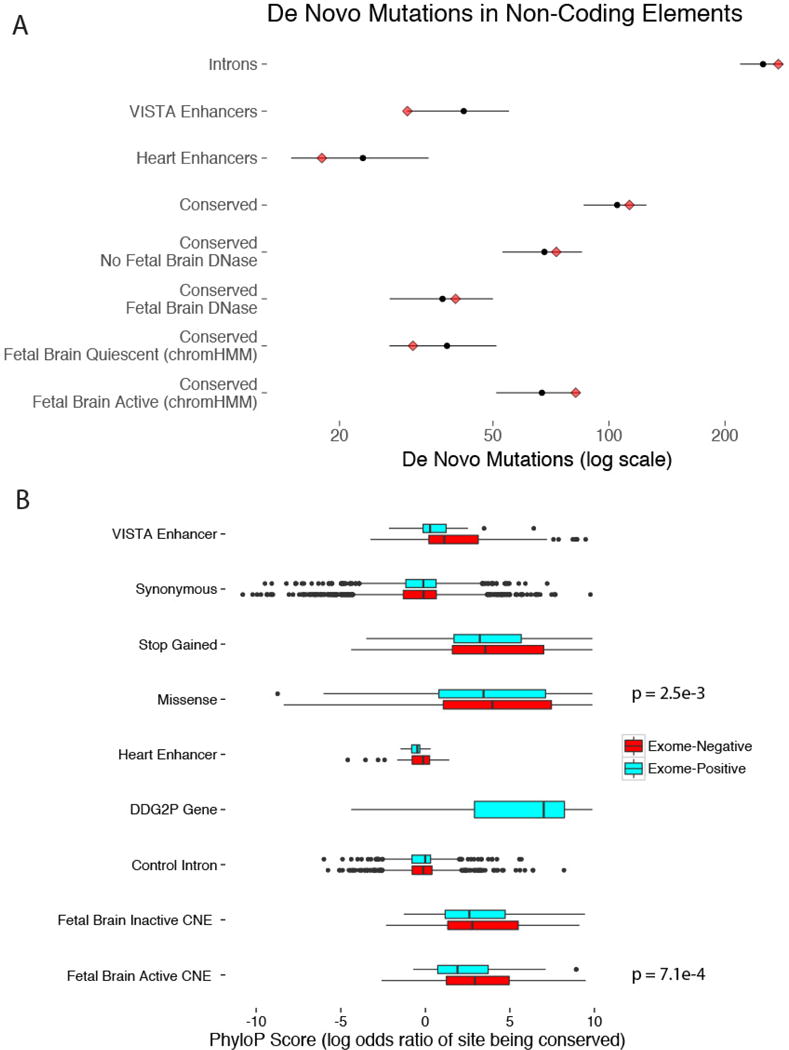
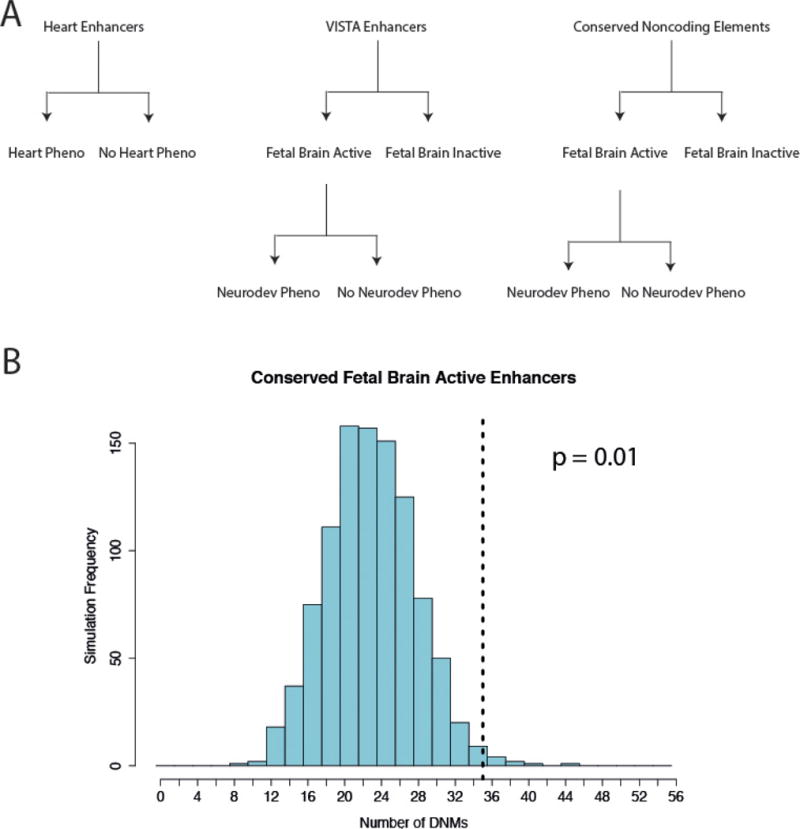
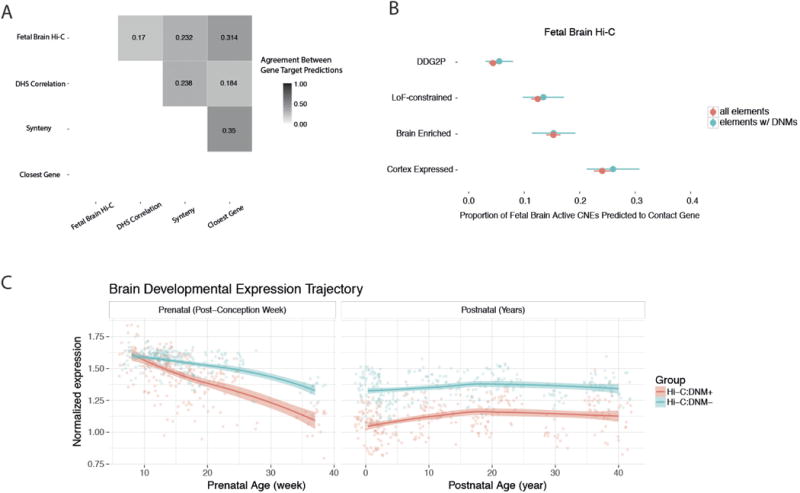
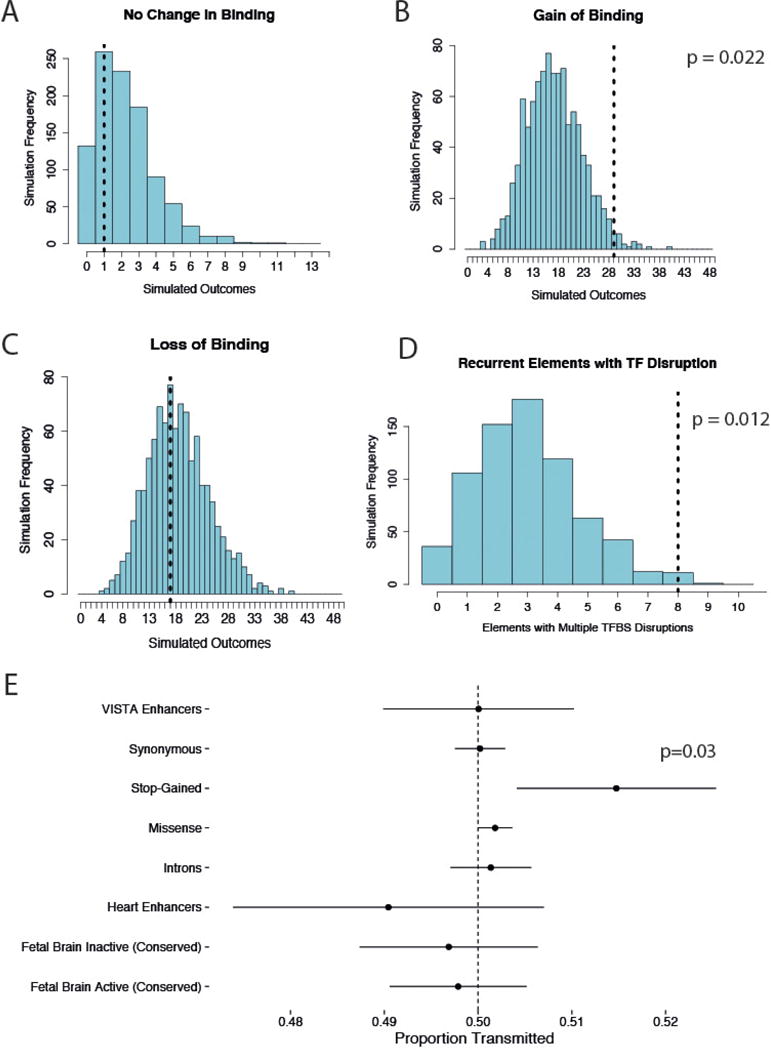
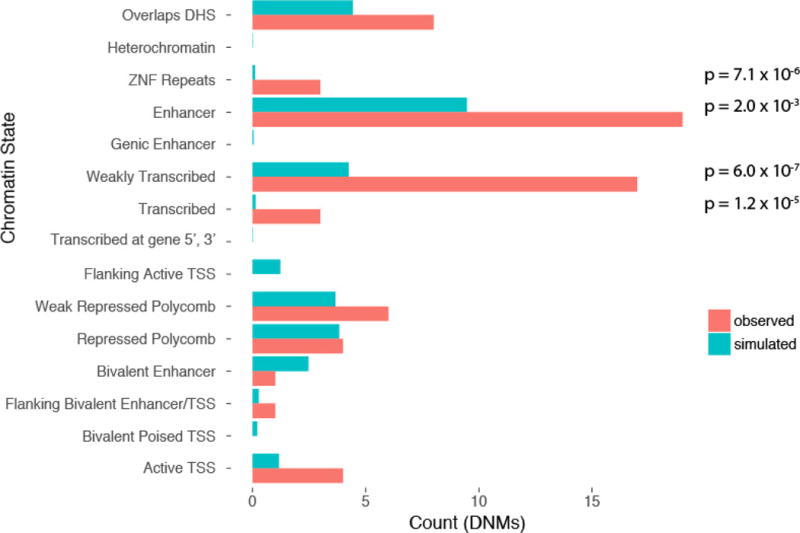
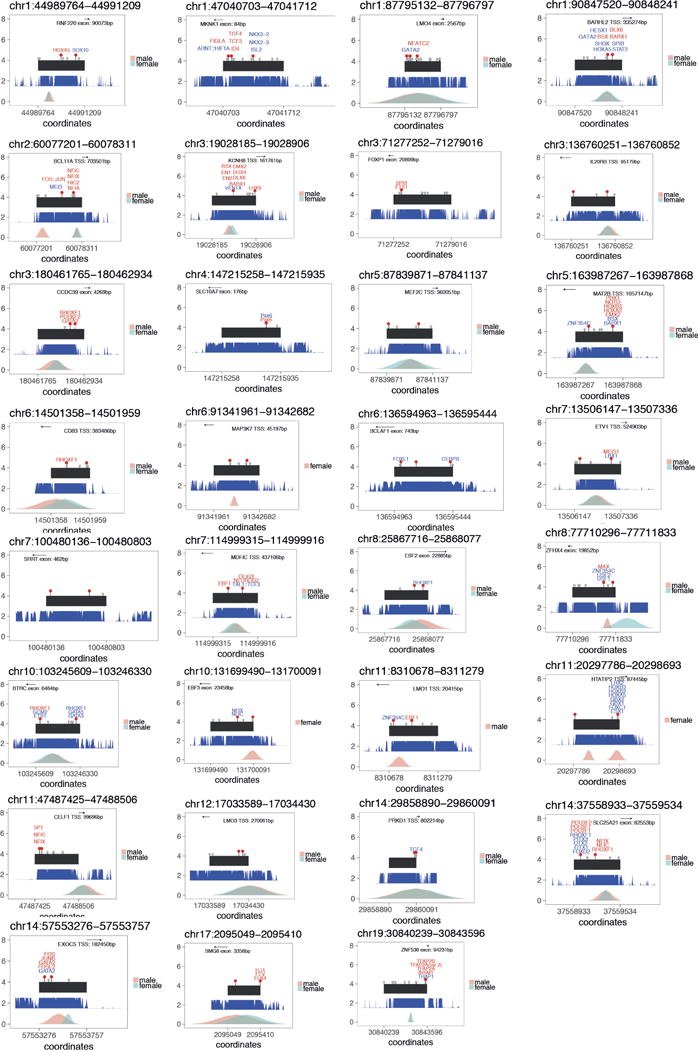
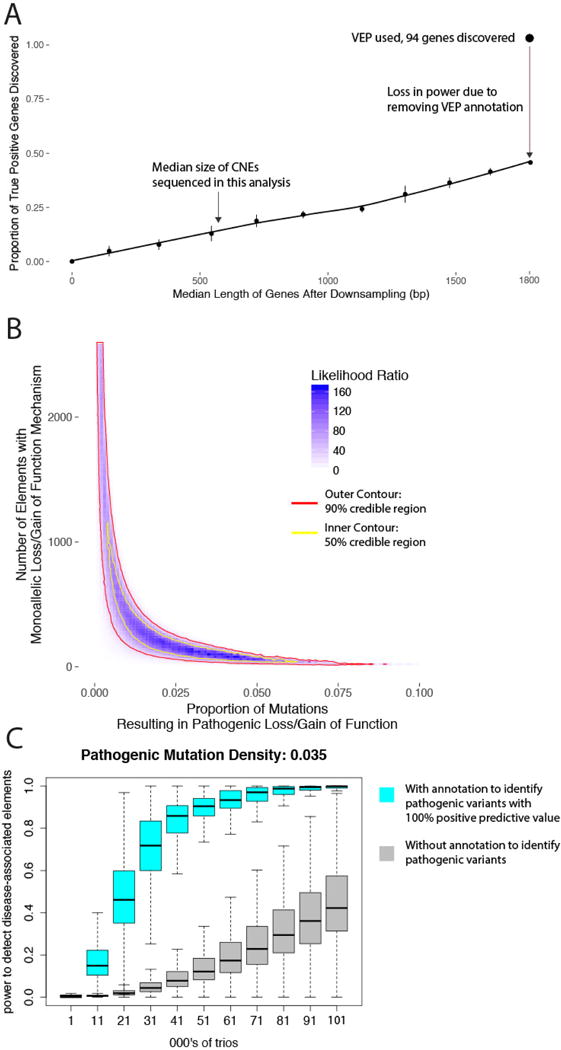
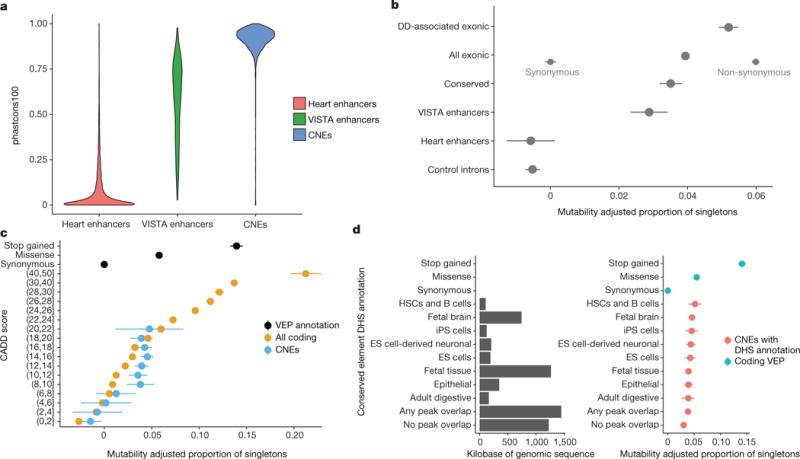
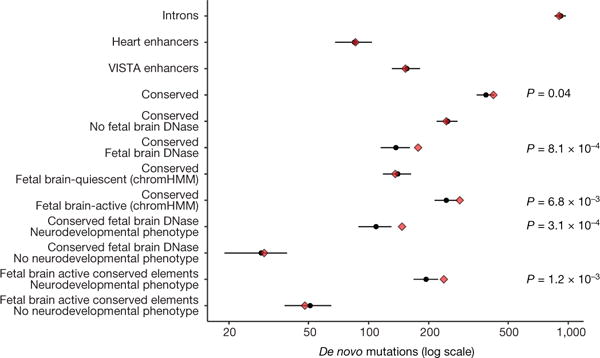
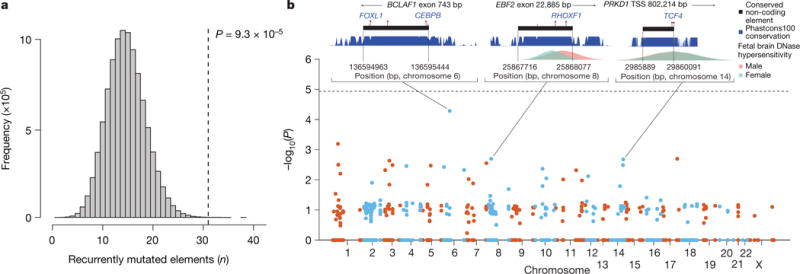
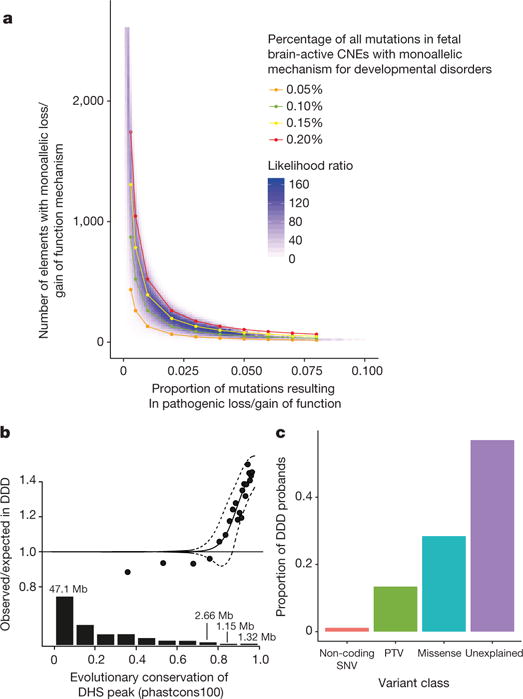
We previously estimated that 42% of patients with severe developmental disorders carry pathogenic de novo mutations in coding sequences. The role of de novo mutations in regulatory elements affecting genes associated with developmental disorders, or other genes, has been essentially unexplored. We identified de novo mutations in three classes of putative regulatory elements in almost 8,000 patients with developmental disorders. Here we show that de novo mutations in highly evolutionarily conserved fetal brain-active elements are significantly and specifically enriched in neurodevelopmental disorders. We identified a significant twofold enrichment of recurrently mutated elements. We estimate that, genome-wide, 1-3% of patients without a diagnostic coding variant carry pathogenic de novo mutations in fetal brain-active regulatory elements and that only 0.15% of all possible mutations within highly conserved fetal brain-active elements cause neurodevelopmental disorders with a dominant mechanism. Our findings represent a robust estimate of the contribution of de novo mutations in regulatory elements to this genetically heterogeneous set of disorders, and emphasize the importance of combining functional and evolutionary evidence to identify regulatory causes of genetic disorders.
Conflict of interest statement
The authors declare competing financial interests: details are available in the online version of the paper. Readers are welcome to comment on the online version of the paper.
Figures