

Trees, fungi and bacteria: tripartite metatranscriptomics of a root microbiome responding to soil contamination
- PMID: 29562928
- PMCID: PMC5863371
- DOI: 10.1186/s40168-018-0432-5
Trees, fungi and bacteria: tripartite metatranscriptomics of a root microbiome responding to soil contamination
Abstract
Background: One method for rejuvenating land polluted with anthropogenic contaminants is through phytoremediation, the reclamation of land through the cultivation of specific crops. The capacity for phytoremediation crops, such as Salix spp., to tolerate and even flourish in contaminated soils relies on a highly complex and predominantly cryptic interacting community of microbial life.
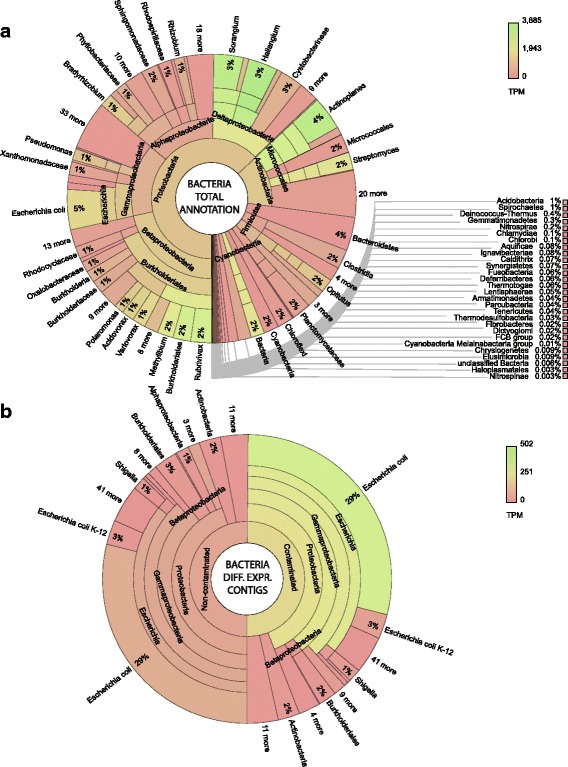
Methods: Here, Illumina HiSeq 2500 sequencing and de novo transcriptome assembly were used to observe gene expression in washed Salix purpurea cv. 'Fish Creek' roots from trees pot grown in petroleum hydrocarbon-contaminated or non-contaminated soil. All 189,849 assembled contigs were annotated without a priori assumption as to sequence origin and differential expression was assessed.
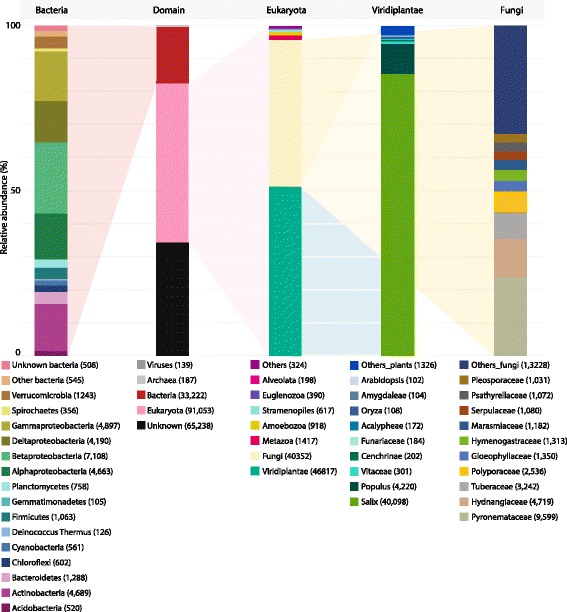
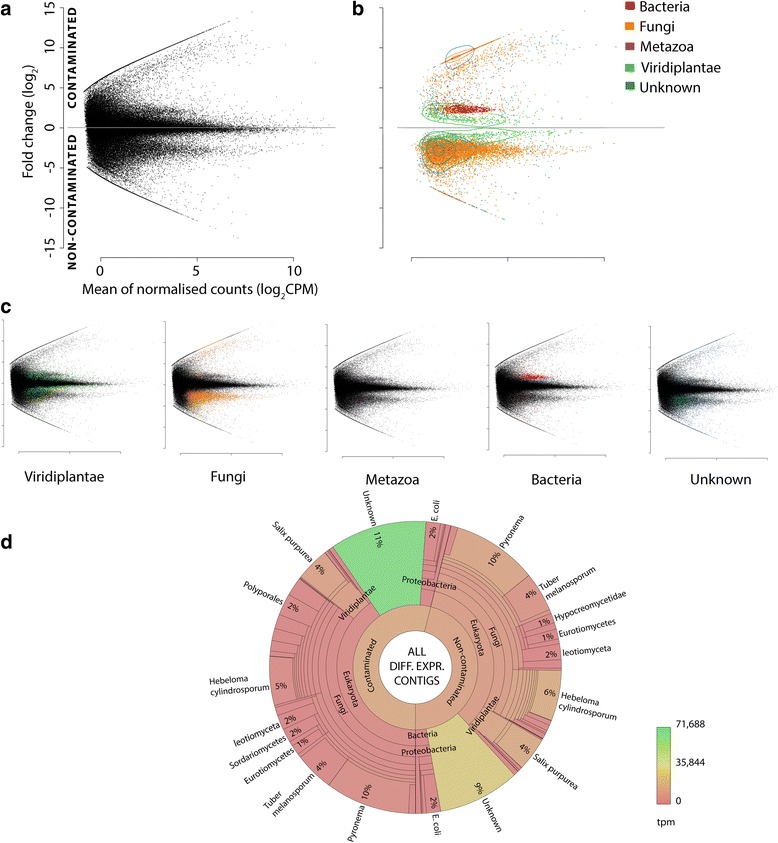
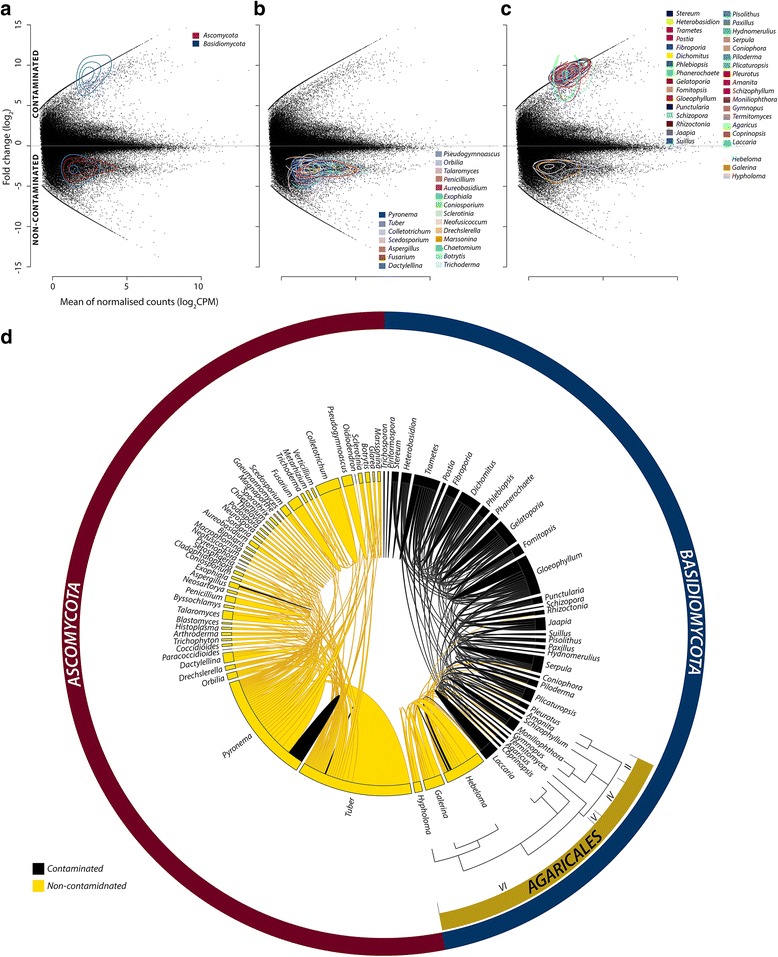
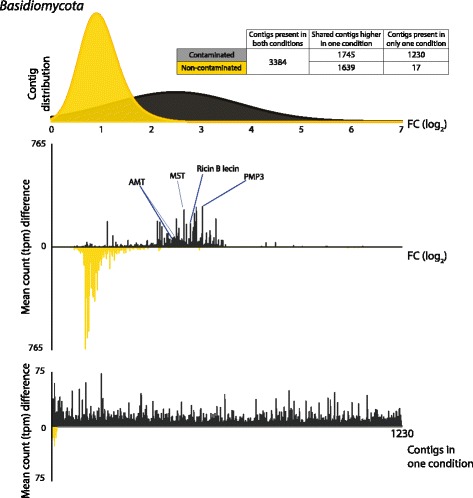
Results: The 839 contigs differentially expressed (DE) and annotated from S. purpurea revealed substantial increases in transcripts encoding abiotic stress response equipment, such as glutathione S-transferases, in roots of contaminated trees as well as the hallmarks of fungal interaction, such as SWEET2 (Sugars Will Eventually Be Exported Transporter). A total of 8252 DE transcripts were fungal in origin, with contamination conditions resulting in a community shift from Ascomycota to Basidiomycota genera. In response to contamination, 1745 Basidiomycota transcripts increased in abundance (the majority uniquely expressed in contaminated soil) including major monosaccharide transporter MST1, primary cell wall and lamella CAZy enzymes, and an ectomycorrhiza-upregulated exo-β-1,3-glucanase (GH5). Additionally, 639 DE polycistronic transcripts from an uncharacterised Enterobacteriaceae species were uniformly in higher abundance in contamination conditions and comprised a wide spectrum of genes cryptic under laboratory conditions but considered putatively involved in eukaryotic interaction, biofilm formation and dioxygenase hydrocarbon degradation.
Conclusions: Fungal gene expression, representing the majority of contigs assembled, suggests out-competition of white rot Ascomycota genera (dominated by Pyronema), a sometimes ectomycorrhizal (ECM) Ascomycota (Tuber) and ECM Basidiomycota (Hebeloma) by a poorly characterised putative ECM Basidiomycota due to contamination. Root and fungal expression involved transcripts encoding carbohydrate/amino acid (C/N) dialogue whereas bacterial gene expression included the apparatus necessary for biofilm interaction and direct reduction of contamination stress, a potential bacterial currency for a role in tripartite mutualism. Unmistakable within the metatranscriptome is the degree to which the landscape of rhizospheric biology, particularly the important but predominantly uncharacterised fungal genetics, is yet to be discovered.
Keywords: Metatranscriptomics; Microbiome; Phytoremediation; Rhizosphere; Salix.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures


References
-
- Brereton NJB, Gonzalez E, Marleau J, Nissim WG, Labrecque M, Joly S, Pitre FE. Comparative transcriptomic approaches exploring contamination stress tolerance in Salix sp. reveal the importance for a metaorganismal de novo assembly approach for nonmodel plants. Plant Physiol. 2016;171:3–24. doi: 10.1104/pp.16.00090. - DOI - PMC - PubMed
-
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. - DOI - PMC - PubMed
-
- Henner P, Schiavon M, Morel JL, Lichtfouse E. Polycyclic aromatic hydrocarbon (PAH) occurrence and remediation methods. Analusis. 1997;25:M56–M59.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
