

Genome-wide determinants of sequence-specific DNA binding of general regulatory factors
- PMID: 29563167
- PMCID: PMC5880240
- DOI: 10.1101/gr.229518.117
Genome-wide determinants of sequence-specific DNA binding of general regulatory factors
Abstract
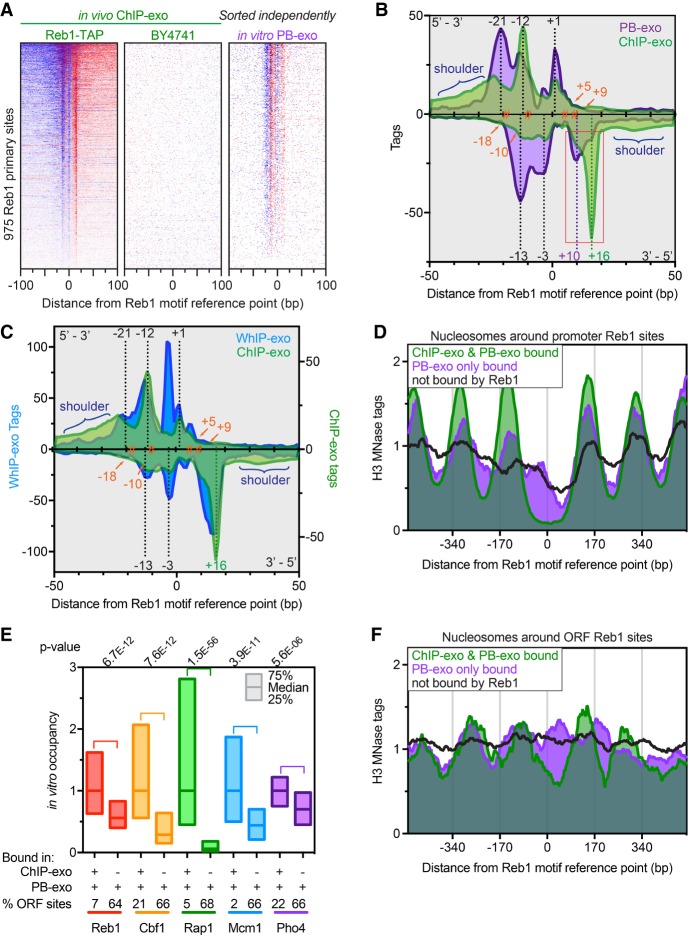
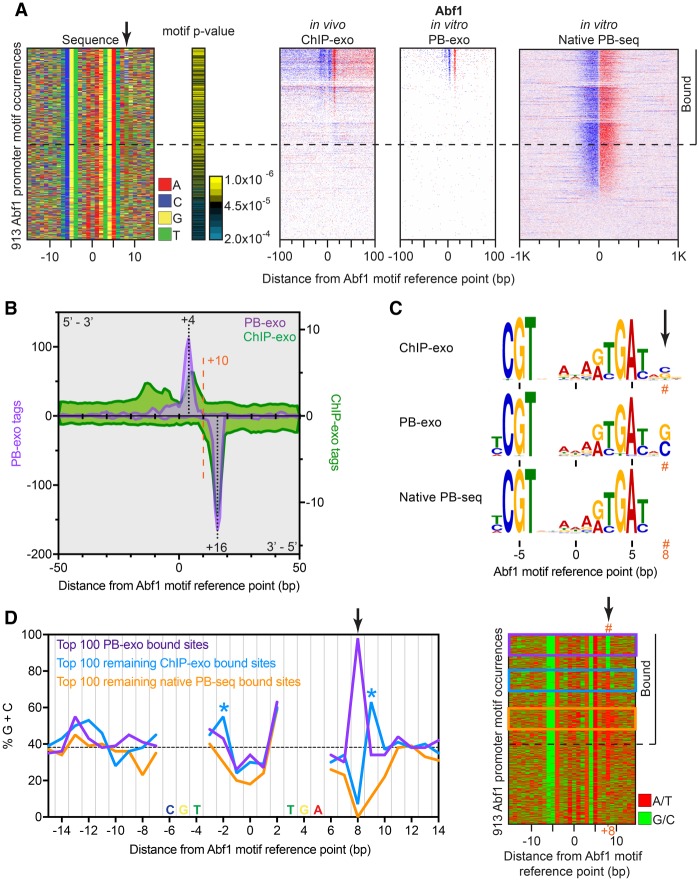
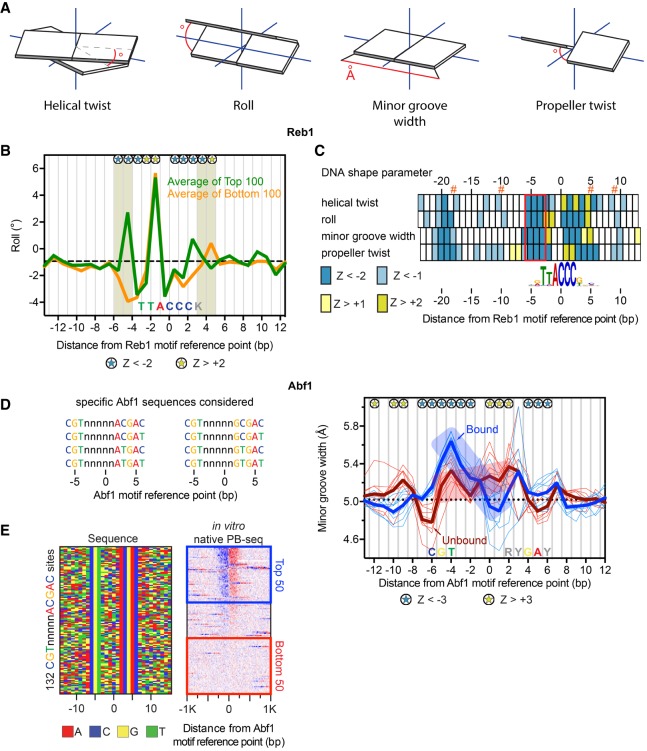
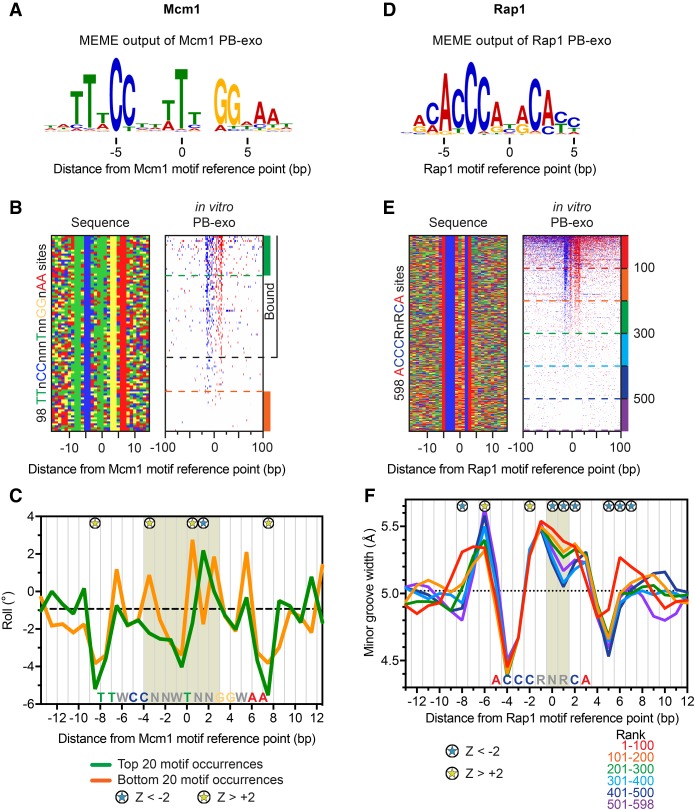
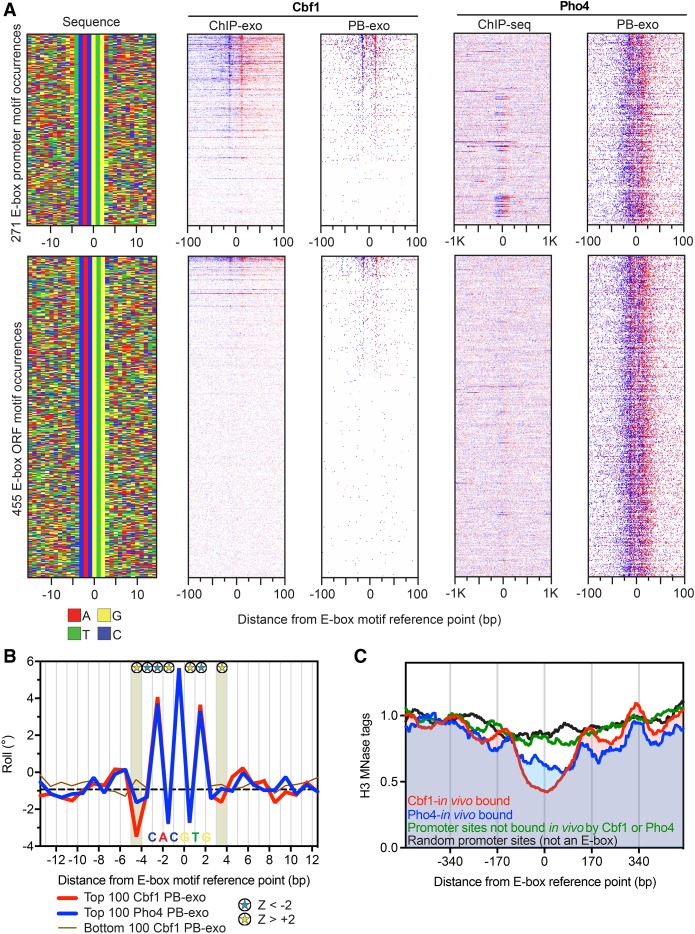
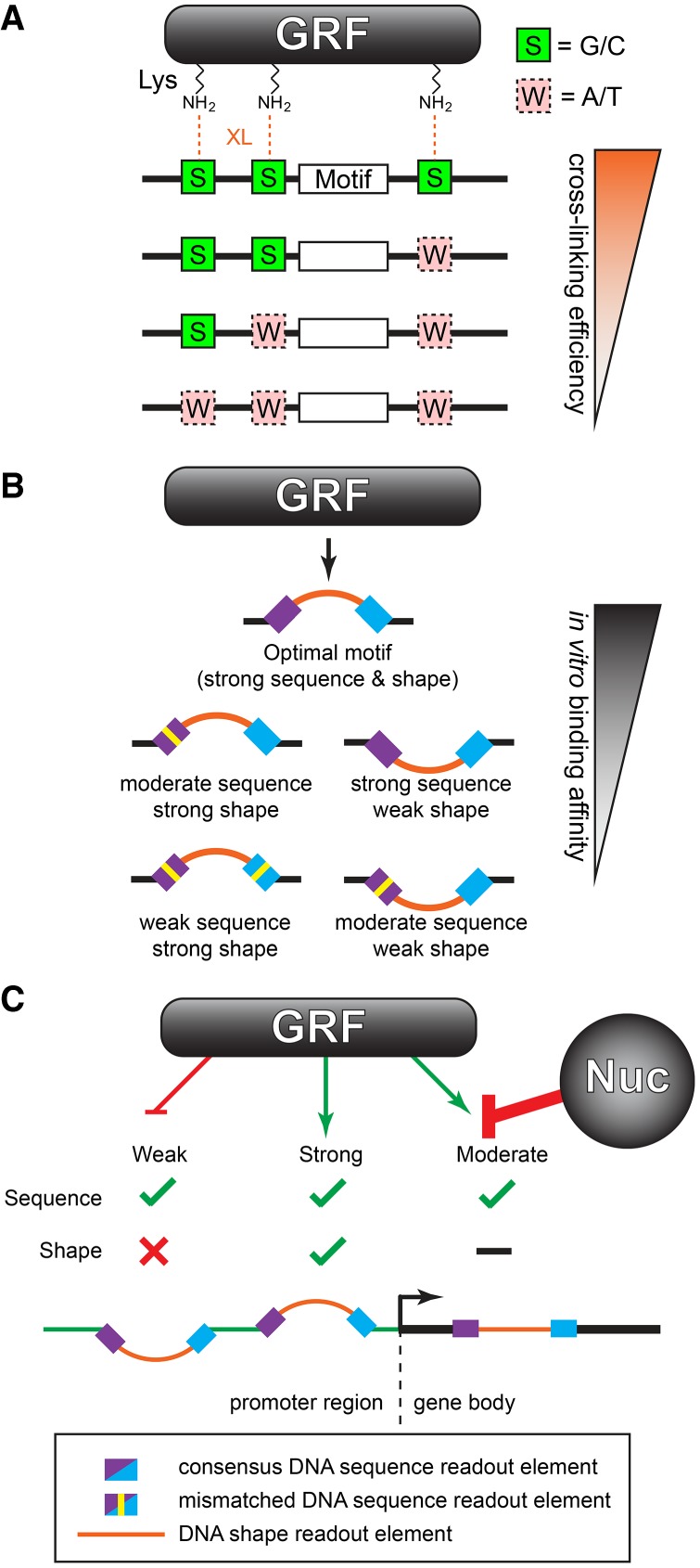
General regulatory factors (GRFs), such as Reb1, Abf1, Rap1, Mcm1, and Cbf1, positionally organize yeast chromatin through interactions with a core consensus DNA sequence. It is assumed that sequence recognition via direct base readout suffices for specificity and that spurious nonfunctional sites are rendered inaccessible by chromatin. We tested these assumptions through genome-wide mapping of GRFs in vivo and in purified biochemical systems at near-base pair (bp) resolution using several ChIP-exo-based assays. We find that computationally predicted DNA shape features (e.g., minor groove width, helix twist, base roll, and propeller twist) that are not defined by a unique consensus sequence are embedded in the nonunique portions of GRF motifs and contribute critically to sequence-specific binding. This dual source specificity occurs at GRF sites in promoter regions where chromatin organization starts. Outside of promoter regions, strong consensus sites lack the shape component and consequently lack an intrinsic ability to bind cognate GRFs, without regard to influences from chromatin. However, sites having a weak consensus and low intrinsic affinity do exist in these regions but are rendered inaccessible in a chromatin environment. Thus, GRF site-specificity is achieved through integration of favorable DNA sequence and shape readouts in promoter regions and by chromatin-based exclusion from fortuitous weak sites within gene bodies. This study further revealed a severe G/C nucleotide cross-linking selectivity inherent in all formaldehyde-based ChIP assays, which includes ChIP-seq. However, for most tested proteins, G/C selectivity did not appreciably affect binding site detection, although it does place limits on the quantitativeness of occupancy levels.
© 2018 Rossi et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous