

Allele-specific differences in transcriptome, miRNome, and mitochondrial function in two hypertrophic cardiomyopathy mouse models
- PMID: 29563334
- PMCID: PMC5926940
- DOI: 10.1172/jci.insight.94493
Allele-specific differences in transcriptome, miRNome, and mitochondrial function in two hypertrophic cardiomyopathy mouse models
Abstract
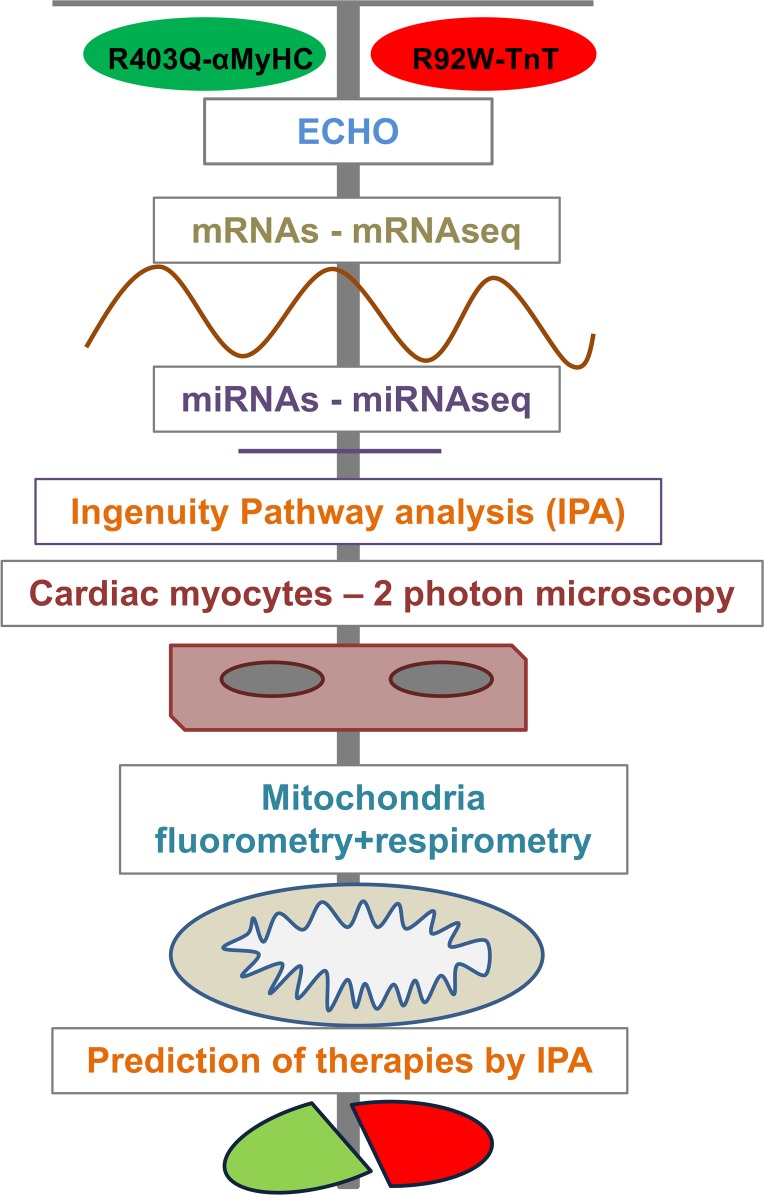
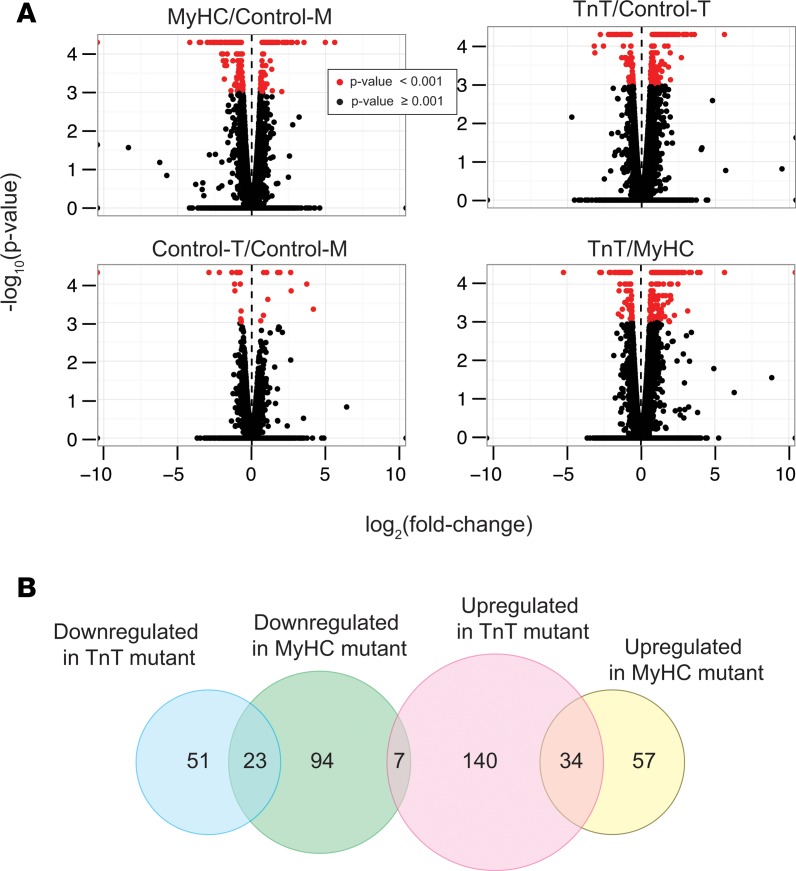
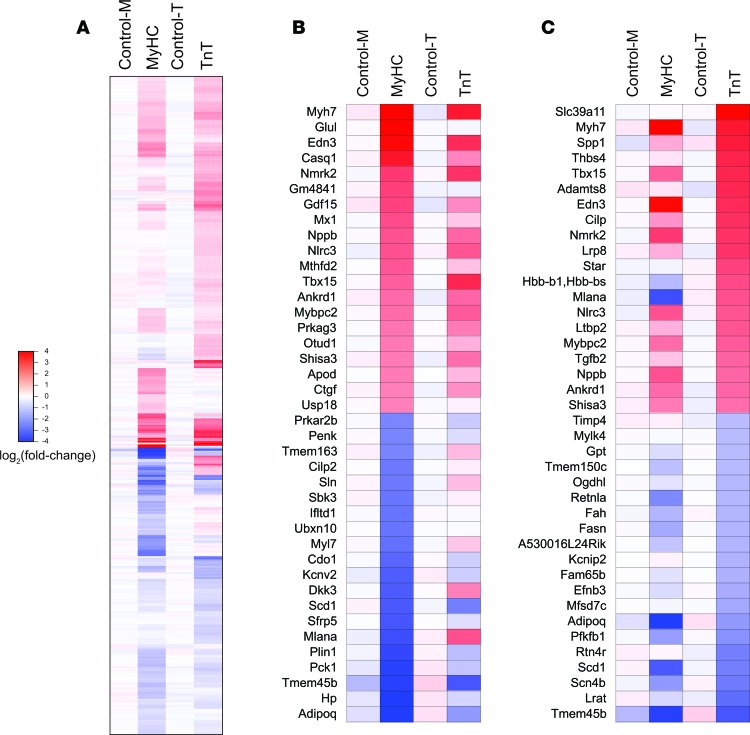
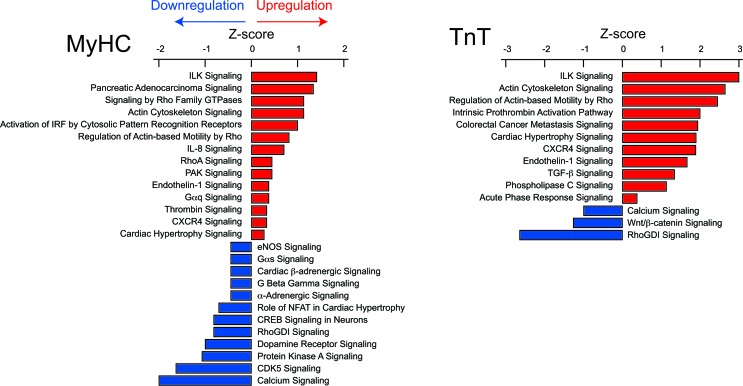
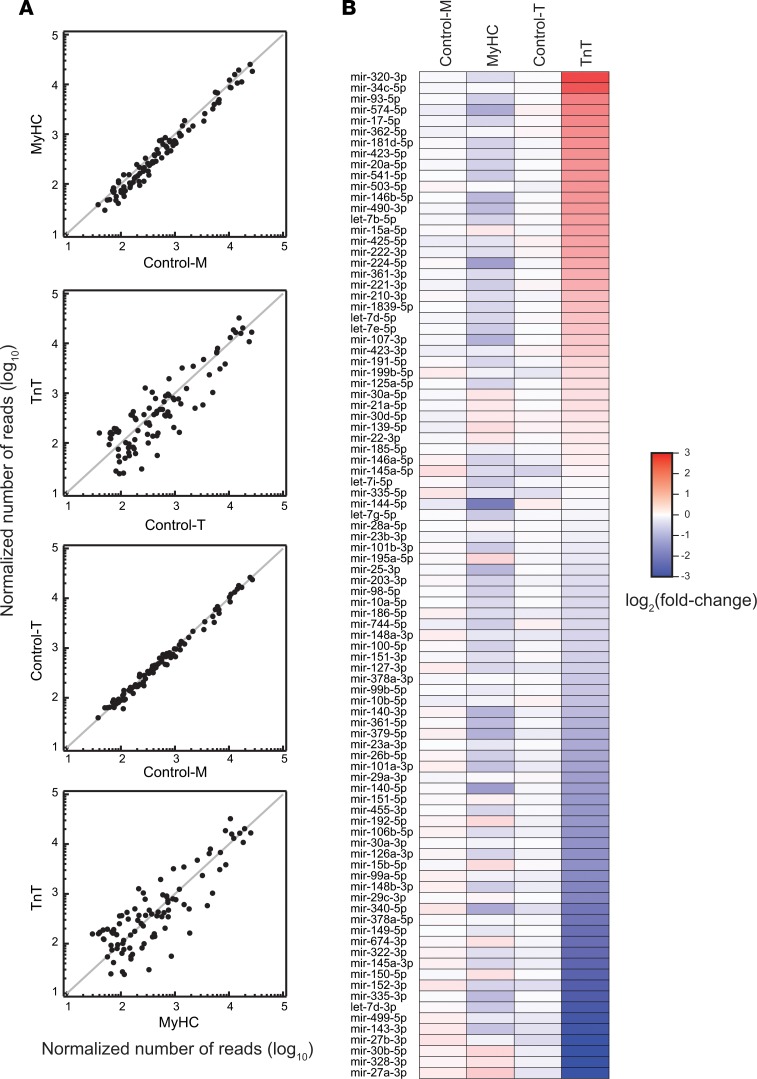
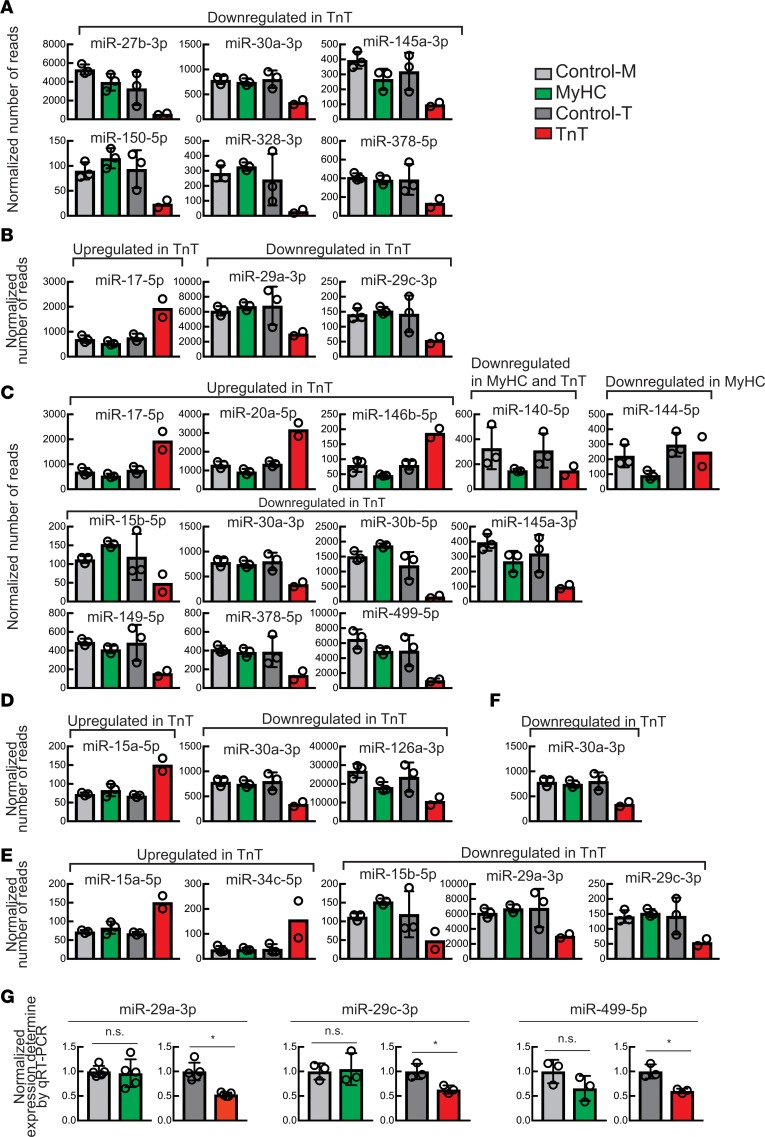
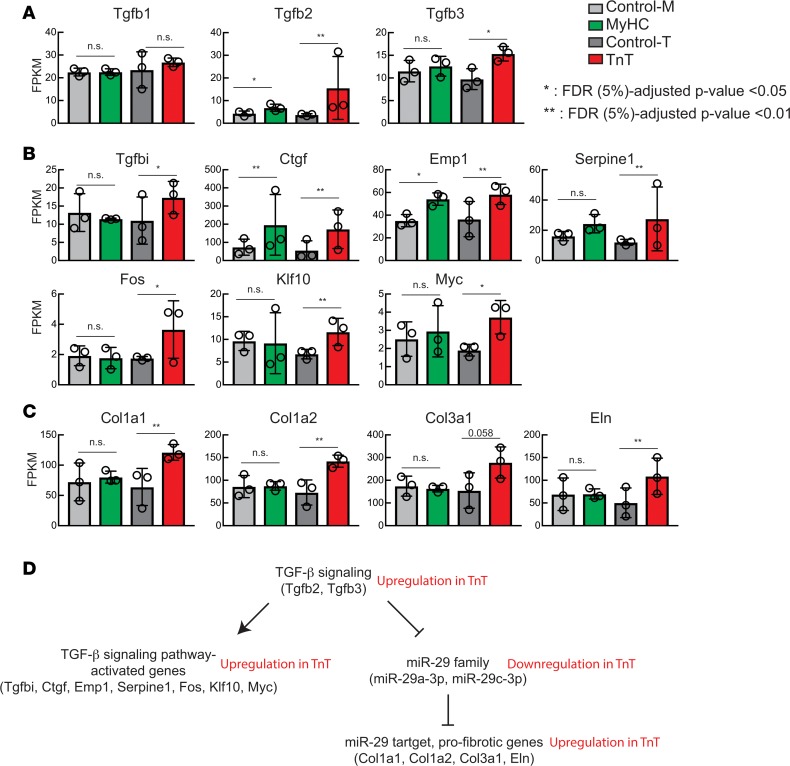
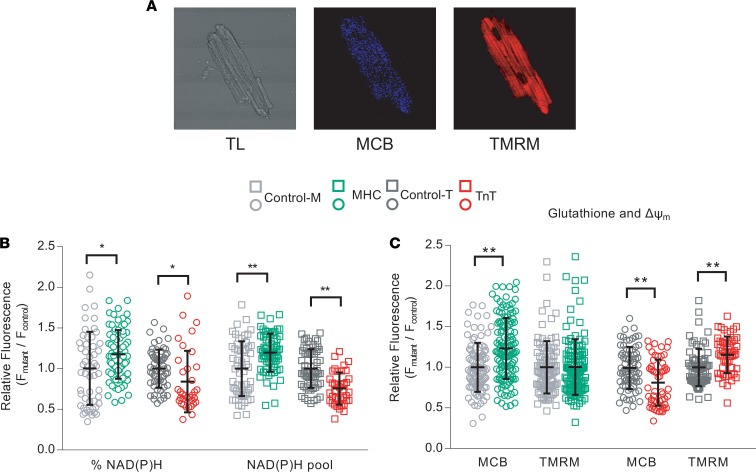
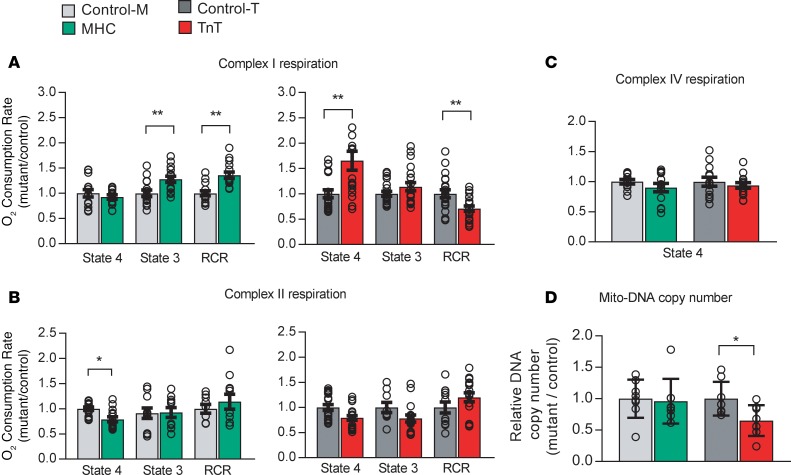
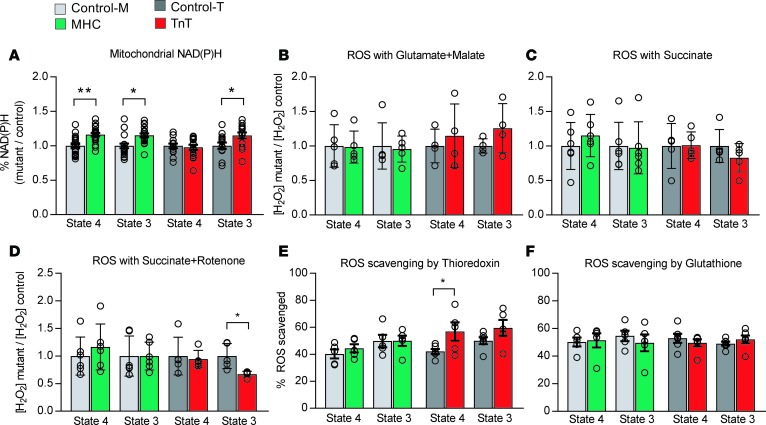
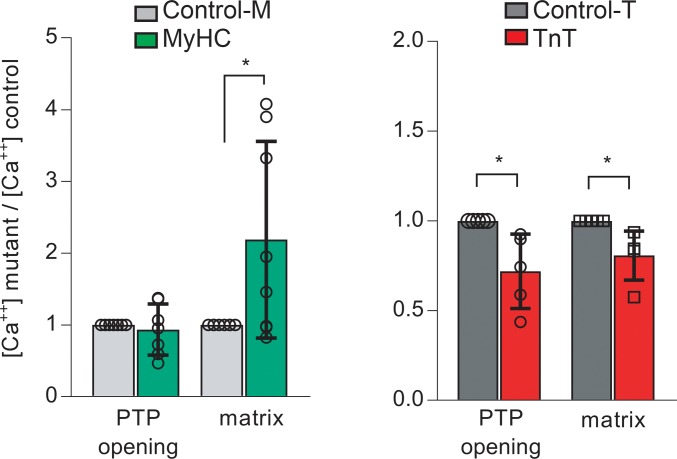
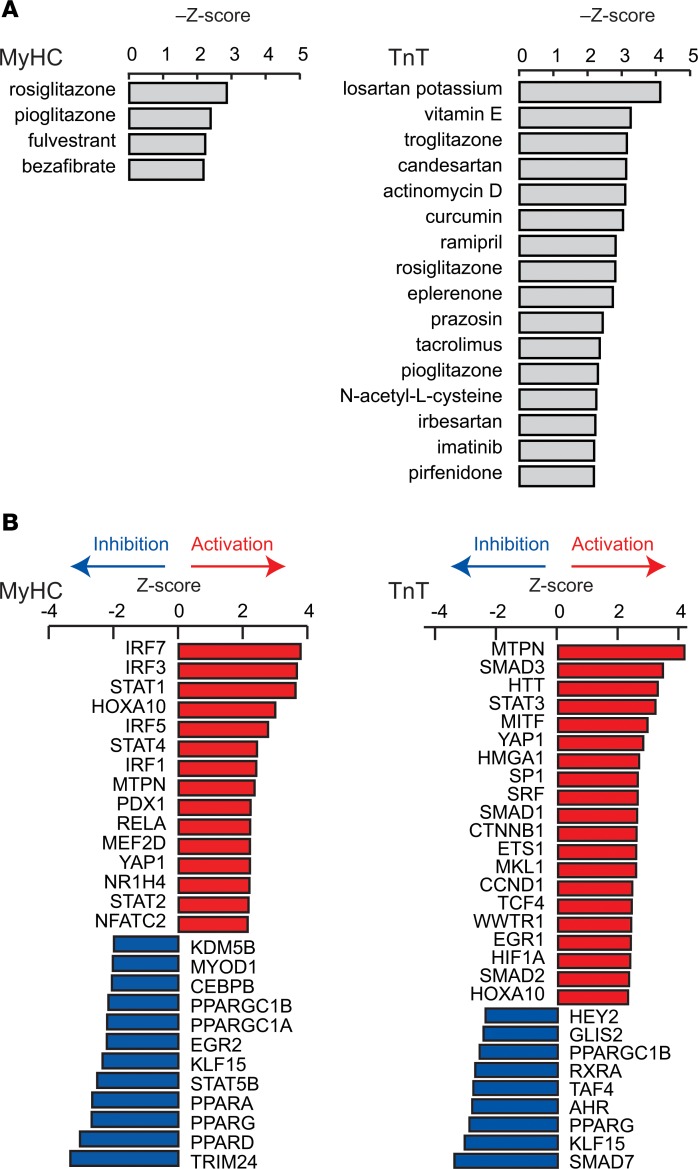
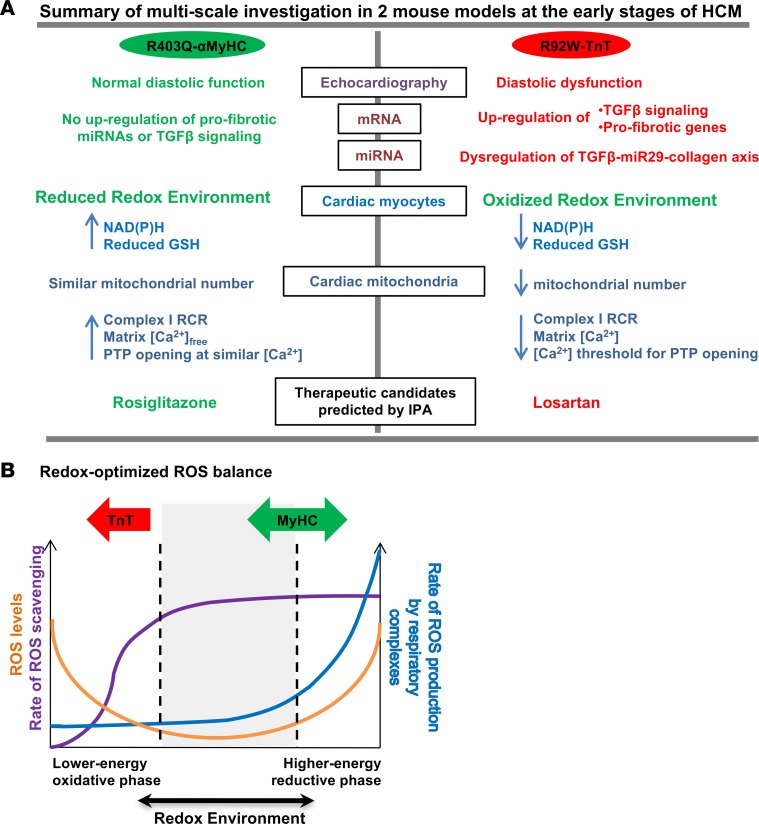
Hypertrophic cardiomyopathy (HCM) stems from mutations in sarcomeric proteins that elicit distinct biophysical sequelae, which in turn may yield radically different intracellular signaling and molecular pathologic profiles. These signaling events remain largely unaddressed by clinical trials that have selected patients based on clinical HCM diagnosis, irrespective of genotype. In this study, we determined how two mouse models of HCM differ, with respect to cellular/mitochondrial function and molecular biosignatures, at an early stage of disease. We show that hearts from young R92W-TnT and R403Q-αMyHC mutation-bearing mice differ in their transcriptome, miRNome, intracellular redox environment, mitochondrial antioxidant defense mechanisms, and susceptibility to mitochondrial permeability transition pore opening. Pathway analysis of mRNA-sequencing data and microRNA profiles indicate that R92W-TnT mutants exhibit a biosignature consistent with activation of profibrotic TGF-β signaling. Our results suggest that the oxidative environment and mitochondrial impairment in young R92W-TnT mice promote activation of TGF-β signaling that foreshadows a pernicious phenotype in young individuals. Of the two mutations, R92W-TnT is more likely to benefit from anti-TGF-β signaling effects conferred by angiotensin receptor blockers and may be responsive to mitochondrial antioxidant strategies in the early stage of disease. Molecular and functional profiling may therefore serve as aids to guide precision therapy for HCM.
Keywords: Cardiology; Cardiovascular disease; Cell Biology; Mitochondria; Transcription.
Conflict of interest statement
Figures
References
-
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287(10):1308–1320. - PubMed
-
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92(4):785–789. doi: 10.1161/01.CIR.92.4.785. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
