

A multiple myeloma-specific capture sequencing platform discovers novel translocations and frequent, risk-associated point mutations in IGLL5
- PMID: 29563506
- PMCID: PMC5862875
- DOI: 10.1038/s41408-018-0062-y
A multiple myeloma-specific capture sequencing platform discovers novel translocations and frequent, risk-associated point mutations in IGLL5
Abstract
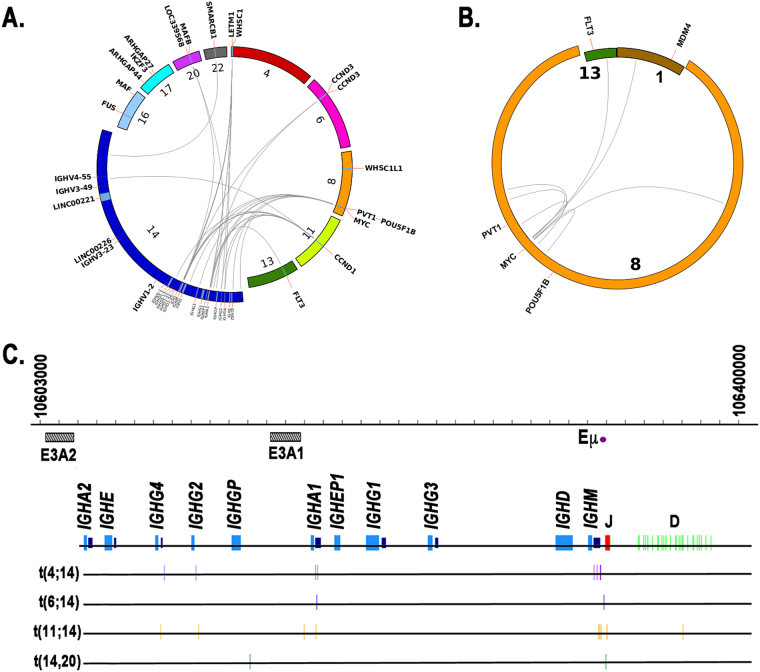
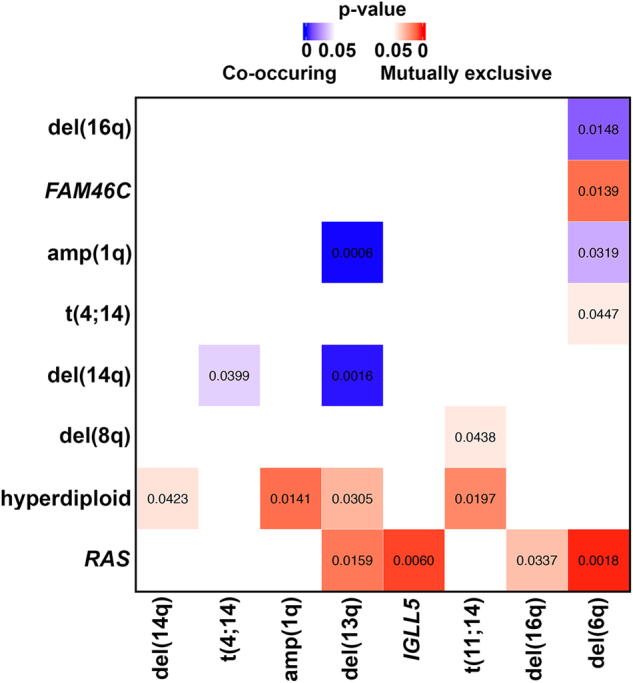
Multiple myeloma (MM) is a disease of copy number variants (CNVs), chromosomal translocations, and single-nucleotide variants (SNVs). To enable integrative studies across these diverse mutation types, we developed a capture-based sequencing platform to detect their occurrence in 465 genes altered in MM and used it to sequence 95 primary tumor-normal pairs to a mean depth of 104×. We detected cases of hyperdiploidy (23%), deletions of 1p (8%), 6q (21%), 8p (17%), 14q (16%), 16q (22%), and 17p (4%), and amplification of 1q (19%). We also detected IGH and MYC translocations near expected frequencies and non-silent SNVs in NRAS (24%), KRAS (21%), FAM46C (17%), TP53 (9%), DIS3 (9%), and BRAF (3%). We discovered frequent mutations in IGLL5 (18%) that were mutually exclusive of RAS mutations and associated with increased risk of disease progression (p = 0.03), suggesting that IGLL5 may be a stratifying biomarker. We identified novel IGLL5/IGH translocations in two samples. We subjected 15 of the pairs to ultra-deep sequencing (1259×) and found that although depth correlated with number of mutations detected (p = 0.001), depth past ~300× added little. The platform provides cost-effective genomic analysis for research and may be useful in individualizing treatment decisions in clinical settings.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures





References
-
- Manier, S. et al. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 14, 100–103 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
