

Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression
- PMID: 29563510
- PMCID: PMC5862881
- DOI: 10.1038/s41467-018-03573-6
Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression
Abstract
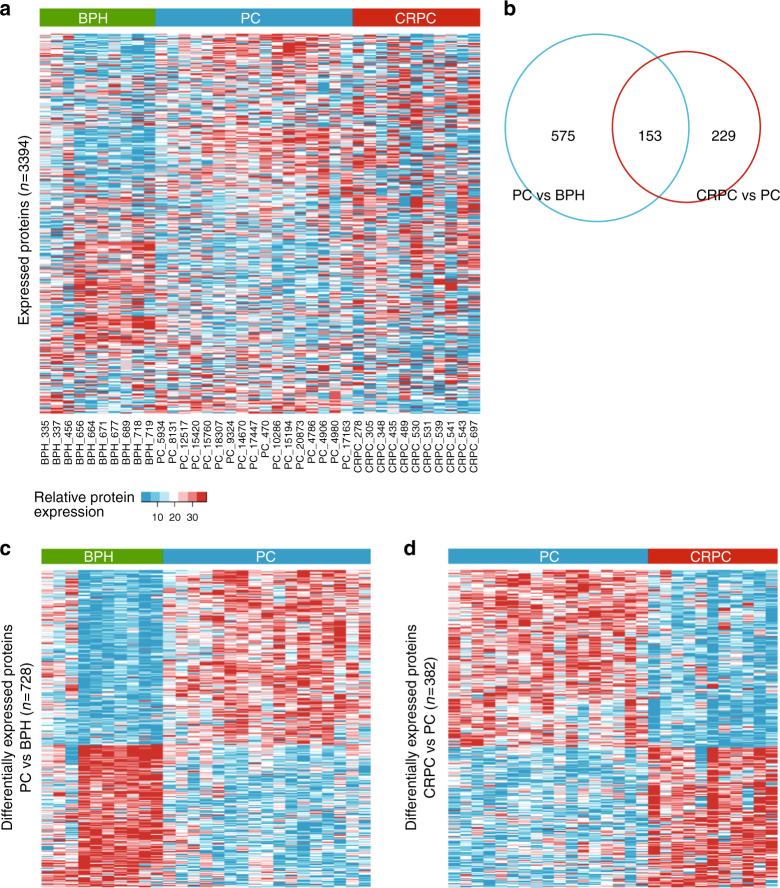
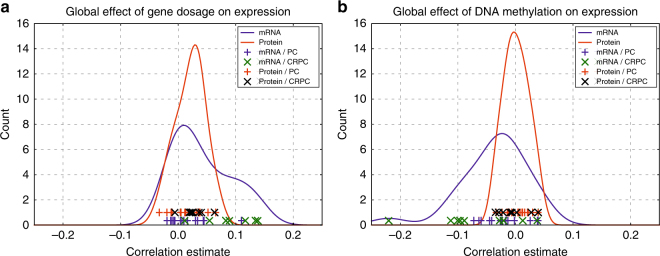
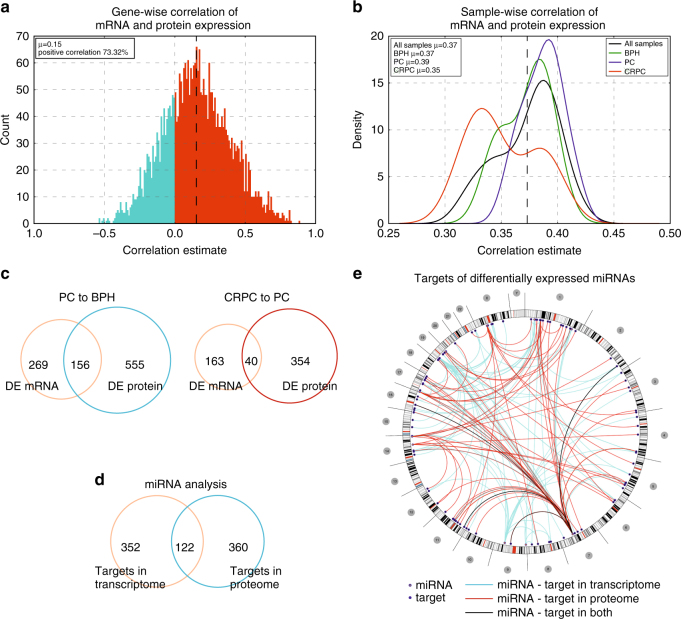
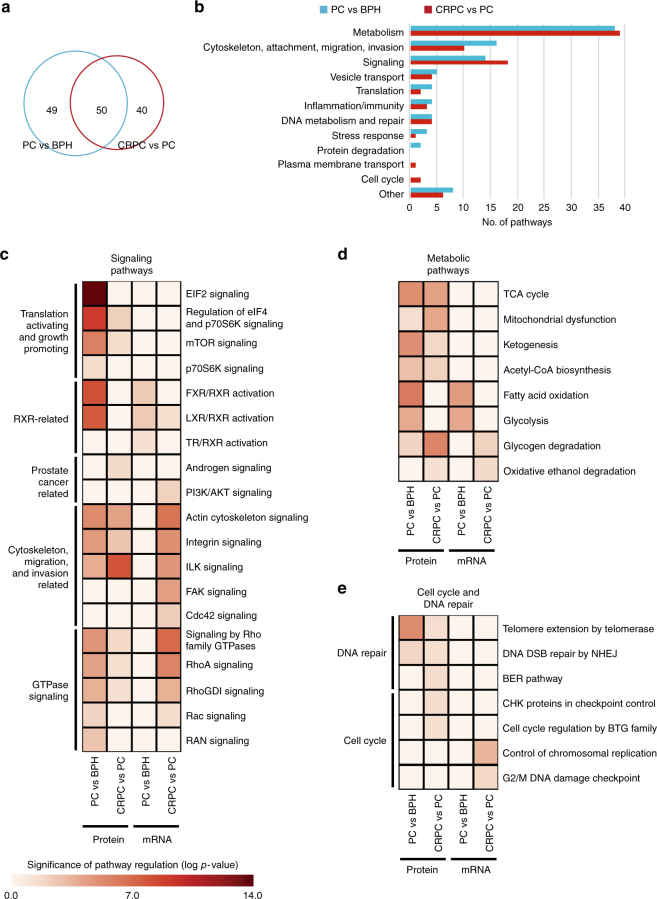
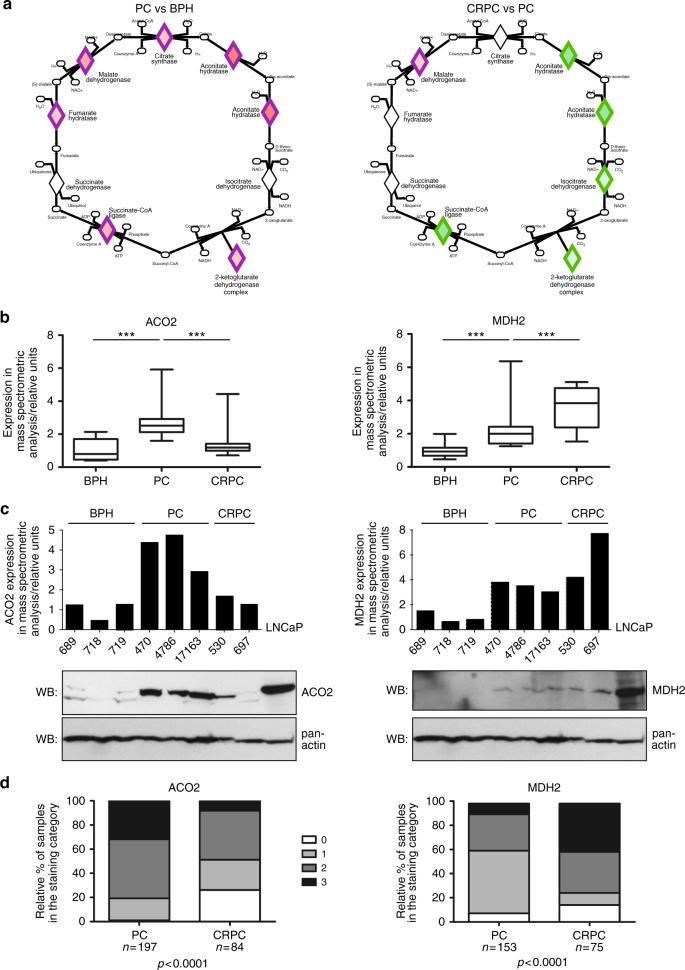
To understand functional consequences of genetic and transcriptional aberrations in prostate cancer, the proteomic changes during disease formation and progression need to be revealed. Here we report high-throughput mass spectrometry on clinical tissue samples of benign prostatic hyperplasia (BPH), untreated primary prostate cancer (PC) and castration resistant prostate cancer (CRPC). Each sample group shows a distinct protein profile. By integrative analysis we show that, especially in CRPC, gene copy number, DNA methylation, and RNA expression levels do not reliably predict proteomic changes. Instead, we uncover previously unrecognized molecular and pathway events, for example, several miRNA target correlations present at protein but not at mRNA level. Notably, we identify two metabolic shifts in the citric acid cycle (TCA cycle) during prostate cancer development and progression. Our proteogenomic analysis uncovers robustness against genomic and transcriptomic aberrations during prostate cancer progression, and significantly extends understanding of prostate cancer disease mechanisms.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Controlling mutational chaos.Nat Rev Urol. 2018 Jun;15(6):335. doi: 10.1038/s41585-018-0021-1. Nat Rev Urol. 2018. PMID: 29743669 No abstract available.
Similar articles
-
Transcriptome Sequencing Reveals PCAT5 as a Novel ERG-Regulated Long Noncoding RNA in Prostate Cancer.Cancer Res. 2015 Oct 1;75(19):4026-31. doi: 10.1158/0008-5472.CAN-15-0217. Epub 2015 Aug 17. Cancer Res. 2015. PMID: 26282172
-
Integrative exploration of the mutual gene signatures and immune microenvironment between benign prostate hyperplasia and castration-resistant prostate cancer.Aging Male. 2023 Dec;26(1):2183947. doi: 10.1080/13685538.2023.2183947. Aging Male. 2023. PMID: 36974949
-
Patient-derived Hormone-naive Prostate Cancer Xenograft Models Reveal Growth Factor Receptor Bound Protein 10 as an Androgen Receptor-repressed Gene Driving the Development of Castration-resistant Prostate Cancer.Eur Urol. 2018 Jun;73(6):949-960. doi: 10.1016/j.eururo.2018.02.019. Epub 2018 Mar 12. Eur Urol. 2018. PMID: 29544736
-
The roles of microRNAs in the progression of castration-resistant prostate cancer.J Hum Genet. 2017 Jan;62(1):25-31. doi: 10.1038/jhg.2016.69. Epub 2016 Jun 9. J Hum Genet. 2017. PMID: 27278789 Review.
-
Non-Coding RNAs in Castration-Resistant Prostate Cancer: Regulation of Androgen Receptor Signaling and Cancer Metabolism.Int J Mol Sci. 2015 Dec 4;16(12):28943-78. doi: 10.3390/ijms161226138. Int J Mol Sci. 2015. PMID: 26690121 Free PMC article. Review.
Cited by
-
Iroquois homeobox 4 (IRX4) derived micropeptide promotes prostate cancer progression and chemoresistance through Wnt signalling dysregulation.Commun Med (Lond). 2024 Nov 1;4(1):224. doi: 10.1038/s43856-024-00613-9. Commun Med (Lond). 2024. PMID: 39487222 Free PMC article.
-
A pathway activity-based proteomic classifier stratifies prostate tumors into two subtypes.Clin Proteomics. 2023 Nov 11;20(1):50. doi: 10.1186/s12014-023-09441-w. Clin Proteomics. 2023. PMID: 37950160 Free PMC article.
-
Metabolic imaging across scales reveals distinct prostate cancer phenotypes.Nat Commun. 2024 Jul 16;15(1):5980. doi: 10.1038/s41467-024-50362-5. Nat Commun. 2024. PMID: 39013948 Free PMC article.
-
Assessing the Potential Prognostic and Immunological Role of TK1 in Prostate Cancer.Front Genet. 2022 Apr 26;13:778850. doi: 10.3389/fgene.2022.778850. eCollection 2022. Front Genet. 2022. PMID: 35559045 Free PMC article.
-
The Proteogenomic Landscape of Curable Prostate Cancer.Cancer Cell. 2019 Mar 18;35(3):414-427.e6. doi: 10.1016/j.ccell.2019.02.005. Cancer Cell. 2019. PMID: 30889379 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
