

A RAS-CaMKKβ-AMPKα2 pathway promotes senescence by licensing post-translational activation of C/EBPβ through a novel 3'UTR mechanism
- PMID: 29563610
- PMCID: PMC6023738
- DOI: 10.1038/s41388-018-0190-7
A RAS-CaMKKβ-AMPKα2 pathway promotes senescence by licensing post-translational activation of C/EBPβ through a novel 3'UTR mechanism
Abstract
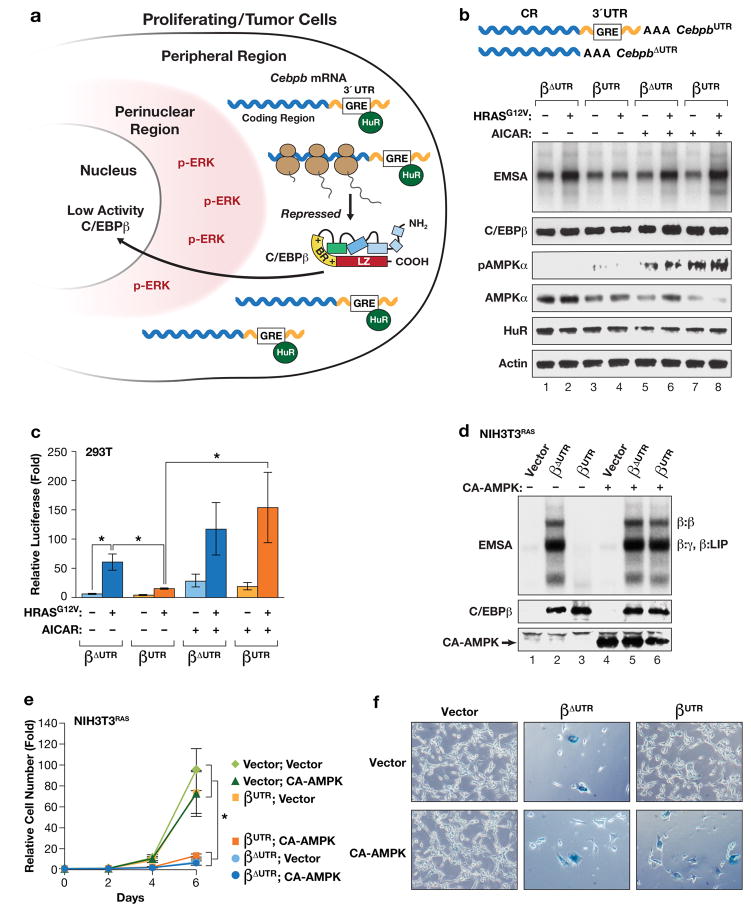
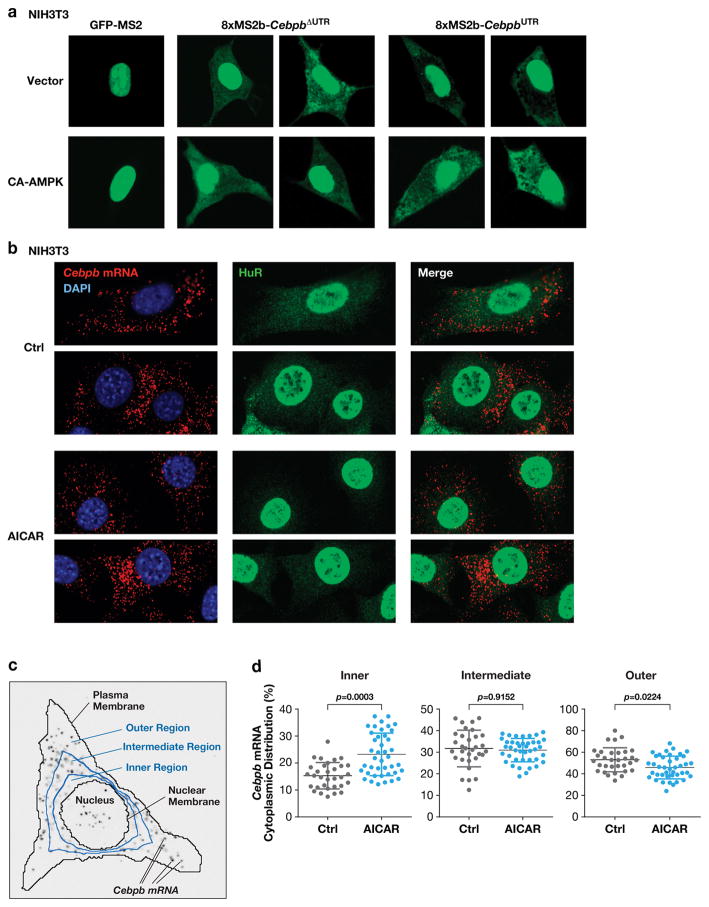
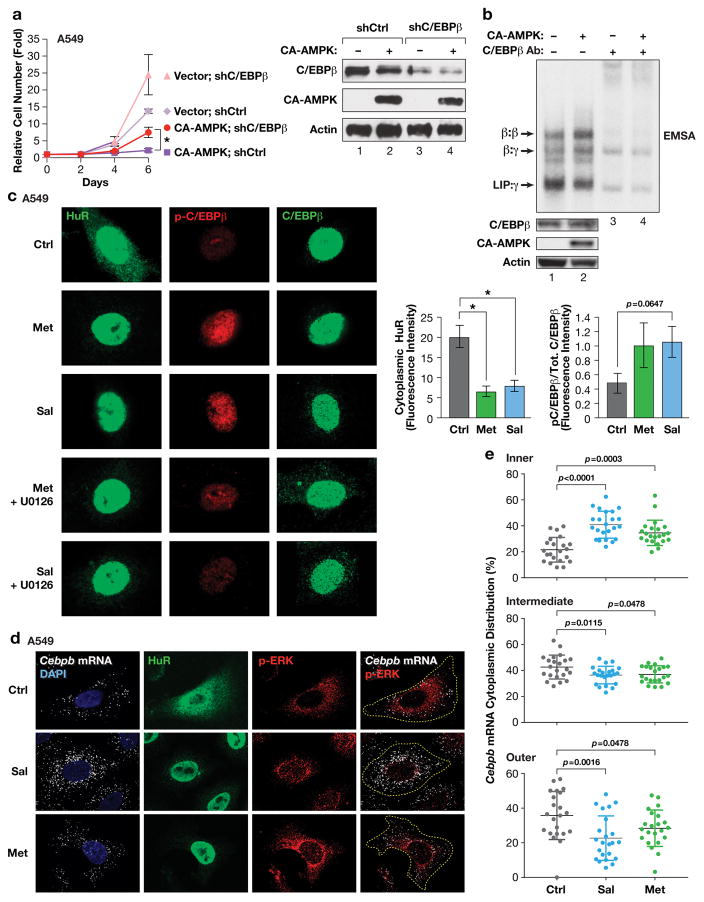
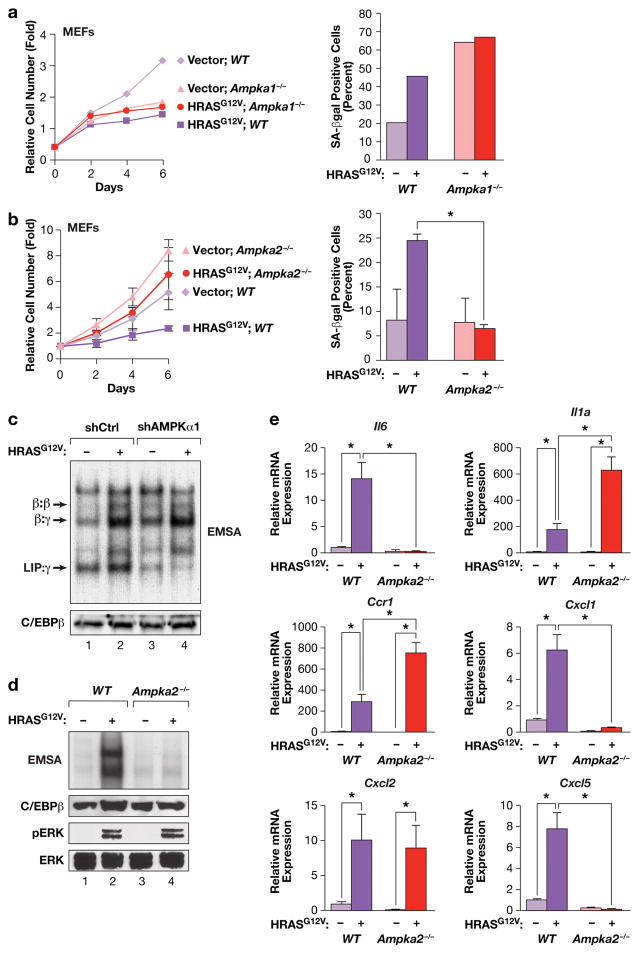
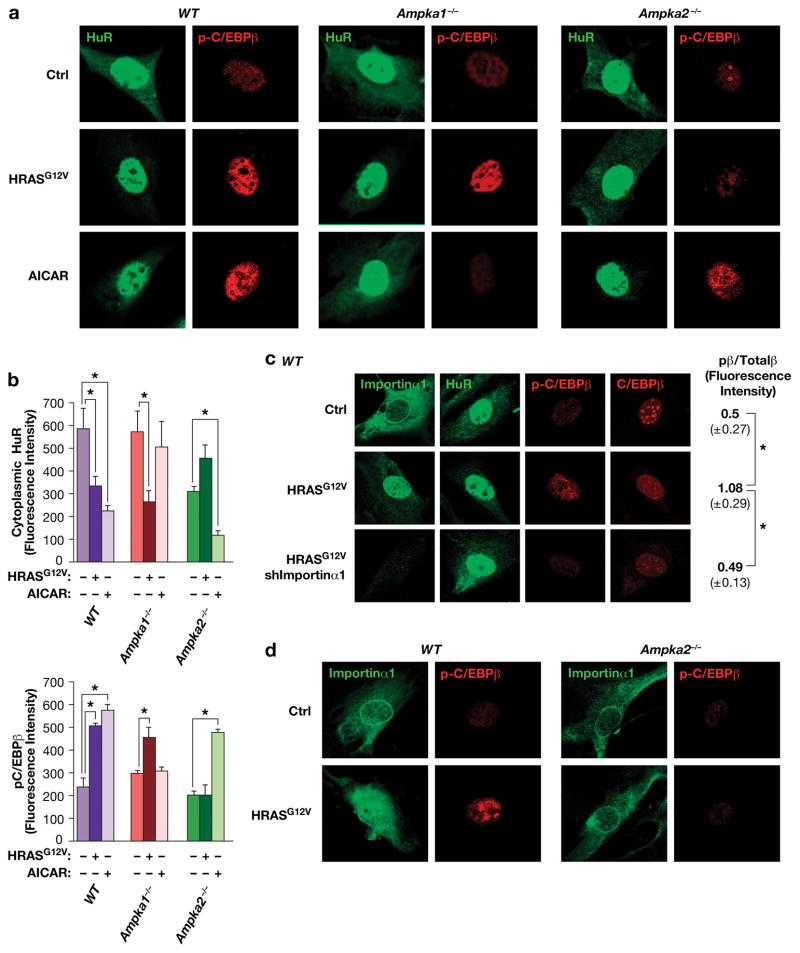
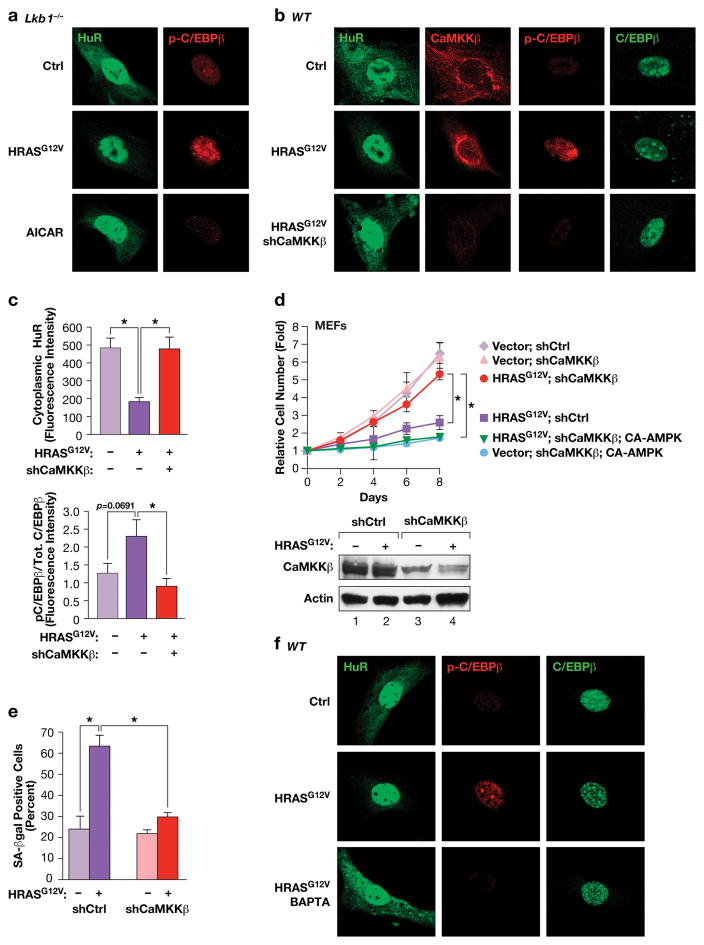
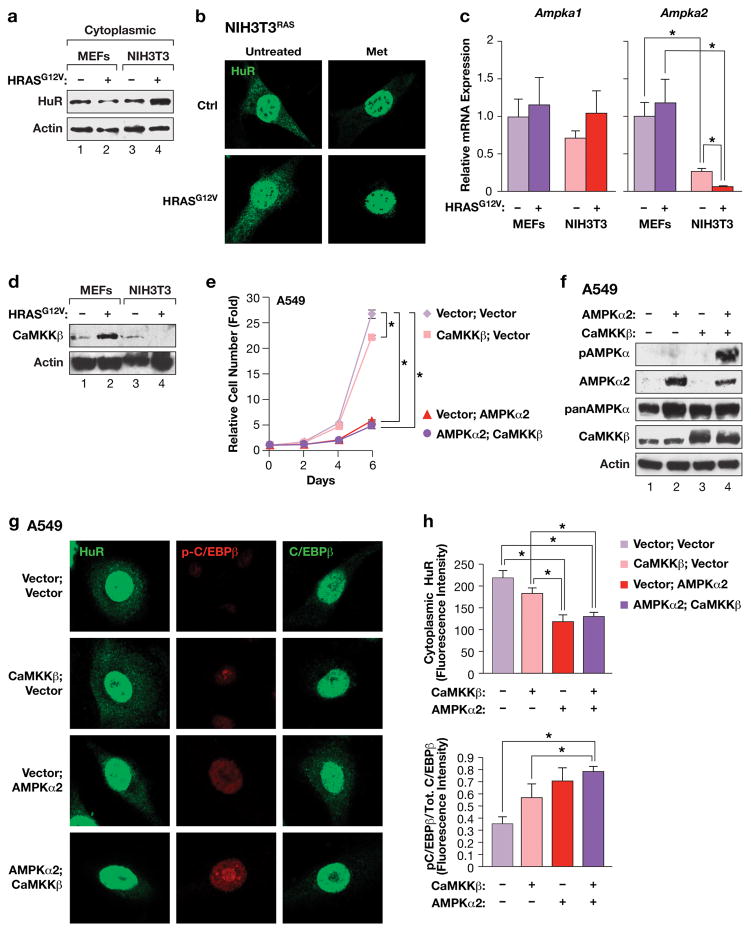
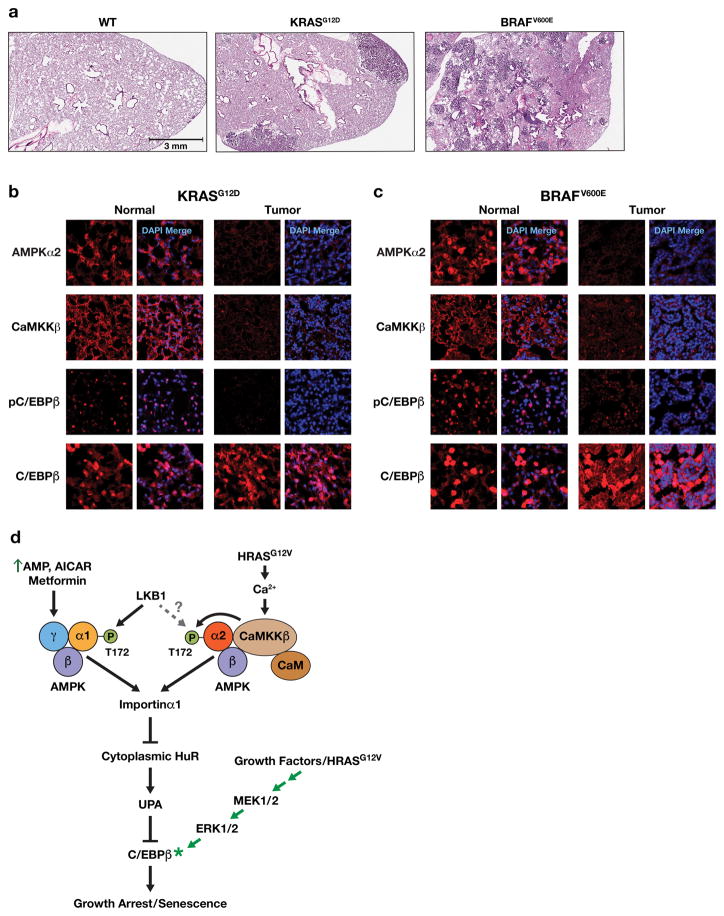
Oncogene-induced senescence (OIS) is an intrinsic tumor suppression mechanism that requires the p53 and RB pathways and post-translational activation of C/EBPβ through the RAS-ERK cascade. We previously reported that in transformed/proliferating cells, C/EBPβ activation is inhibited by G/U-rich elements (GREs) in its 3'UTR. This mechanism, termed "3'UTR regulation of protein activity" (UPA), maintains C/EBPβ in a low-activity state in tumor cells and thus facilitates senescence bypass. Here we show that C/EBPβ UPA is overridden by AMPK signaling. AMPK activators decrease cytoplasmic levels of the GRE binding protein HuR, which is a key UPA component. Reduced cytoplasmic HuR disrupts 3'UTR-mediated trafficking of Cebpb transcripts to the peripheral cytoplasm-a fundamental feature of UPA-thereby stimulating C/EBPβ activation and growth arrest. In primary cells, oncogenic RAS triggers a Ca++-CaMKKβ-AMPKα2-HuR pathway, independent of AMPKα1, that is essential for C/EBPβ activation and OIS. This axis is disrupted in cancer cells through down-regulation of AMPKα2 and CaMKKβ. Thus, CaMKKβ-AMPKα2 signaling constitutes a key tumor suppressor pathway that activates a novel UPA-cancelling mechanism to unmask the cytostatic and pro-senescence functions of C/EBPβ.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
3'UTR elements inhibit Ras-induced C/EBPβ post-translational activation and senescence in tumour cells.EMBO J. 2011 Jul 29;30(18):3714-28. doi: 10.1038/emboj.2011.250. EMBO J. 2011. PMID: 21804532 Free PMC article.
-
RasV12-mediated down-regulation of CCAAT/enhancer binding protein beta in immortalized fibroblasts requires loss of p19Arf and facilitates bypass of oncogene-induced senescence.Cancer Res. 2009 Mar 15;69(6):2588-98. doi: 10.1158/0008-5472.CAN-08-2312. Epub 2009 Mar 10. Cancer Res. 2009. PMID: 19276382 Free PMC article.
-
Oncogenic RAS-Induced Perinuclear Signaling Complexes Requiring KSR1 Regulate Signal Transmission to Downstream Targets.Cancer Res. 2018 Feb 15;78(4):891-908. doi: 10.1158/0008-5472.CAN-17-2353. Epub 2017 Dec 19. Cancer Res. 2018. PMID: 29259016 Free PMC article.
-
Regulation of senescence and the SASP by the transcription factor C/EBPβ.Exp Gerontol. 2019 Dec;128:110752. doi: 10.1016/j.exger.2019.110752. Epub 2019 Oct 22. Exp Gerontol. 2019. PMID: 31648009 Review.
-
Stop and go: anti-proliferative and mitogenic functions of the transcription factor C/EBPbeta.Cell Cycle. 2006 May;5(9):953-7. doi: 10.4161/cc.5.9.2733. Epub 2006 May 1. Cell Cycle. 2006. PMID: 16687924 Review.
Cited by
-
OR2AT4, an Ectopic Olfactory Receptor, Suppresses Oxidative Stress-Induced Senescence in Human Keratinocytes.Antioxidants (Basel). 2022 Nov 3;11(11):2180. doi: 10.3390/antiox11112180. Antioxidants (Basel). 2022. PMID: 36358552 Free PMC article.
-
The RNA-Binding Protein HuR in Digestive System Tumors.Biomed Res Int. 2020 Jul 24;2020:9656051. doi: 10.1155/2020/9656051. eCollection 2020. Biomed Res Int. 2020. PMID: 32775456 Free PMC article. Review.
-
The "Janus" Role of C/EBPs Family Members in Cancer Progression.Int J Mol Sci. 2020 Jun 17;21(12):4308. doi: 10.3390/ijms21124308. Int J Mol Sci. 2020. PMID: 32560326 Free PMC article. Review.
-
AMPK, a key molecule regulating aging-related myocardial ischemia-reperfusion injury.Mol Biol Rep. 2024 Feb 1;51(1):257. doi: 10.1007/s11033-023-09050-8. Mol Biol Rep. 2024. PMID: 38302614 Review.
-
Icaritin induces cellular senescence by accumulating the ROS production and regulation of the Jak2/Stat3/p21 pathway in imatinib-resistant, chronic myeloid leukemia cells.Am J Transl Res. 2021 Aug 15;13(8):8860-8872. eCollection 2021. Am J Transl Res. 2021. PMID: 34540000 Free PMC article.
References
-
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
