

Early MinION™ nanopore single-molecule sequencing technology enables the characterization of hepatitis B virus genetic complexity in clinical samples
- PMID: 29566006
- PMCID: PMC5864009
- DOI: 10.1371/journal.pone.0194366
Early MinION™ nanopore single-molecule sequencing technology enables the characterization of hepatitis B virus genetic complexity in clinical samples
Abstract
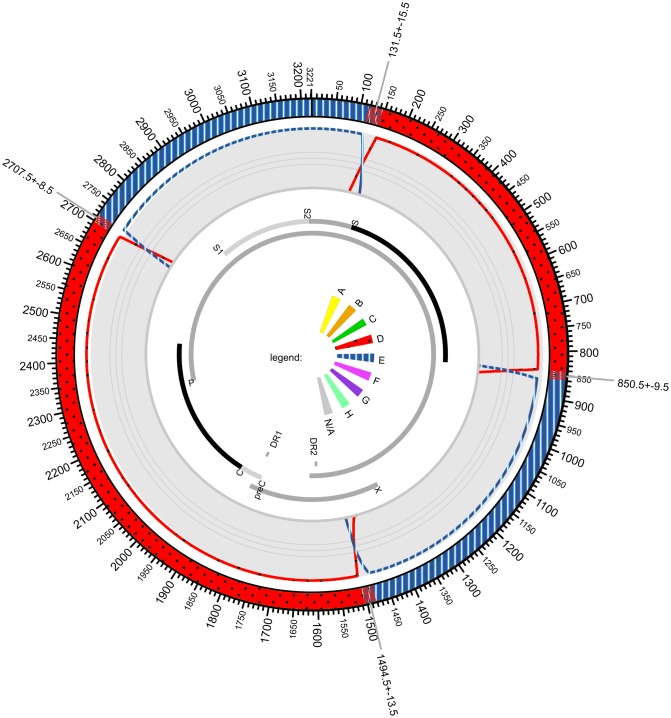
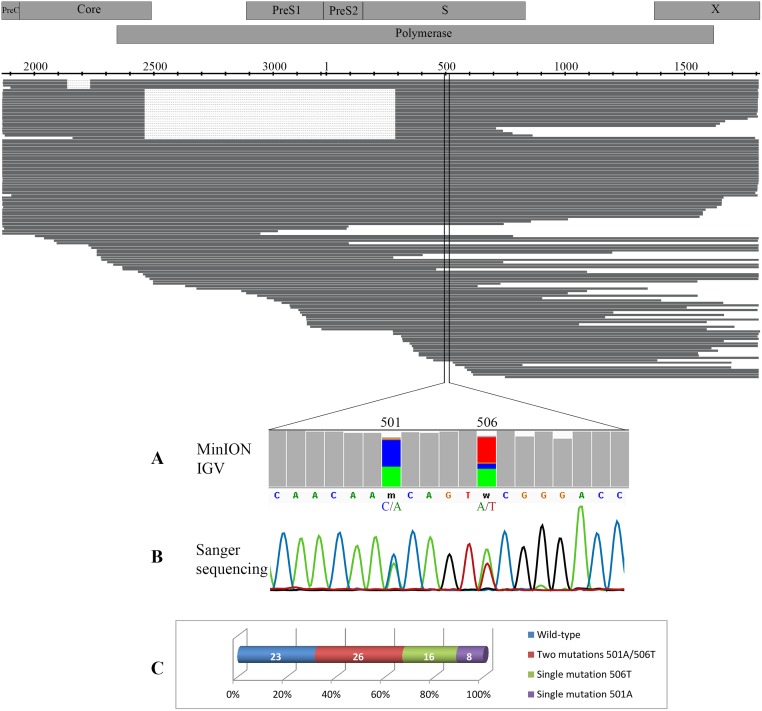
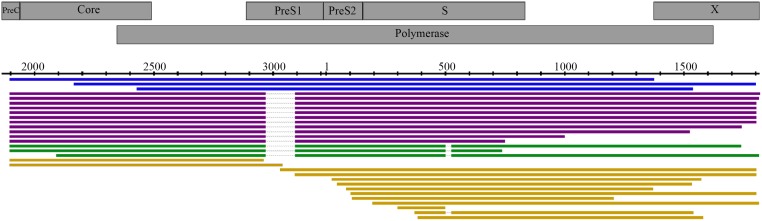
Until recently, the method of choice to characterize viral diversity consisted in cloning PCR amplicons of full-length viral genomes and Sanger-sequencing of multiple clones. However, this is extremely laborious, time-consuming, and low-throughput. Next generation short-read sequencing appears also limited by its inability to directly sequence full-length viral genomes. The MinION™ device recently developed by Oxford Nanopore Technologies can be a promising alternative by applying long-read single-molecule sequencing directly to the overall amplified products generated in a PCR reaction. This new technology was evaluated by using hepatitis B virus (HBV) as a model. Several previously characterized HBV-infected clinical samples were investigated including recombinant virus, variants that harbored deletions and mixed population. Original MinION device was able to generate individual complete 3,200-nt HBV genome sequences and to identify recombinant variants. MinION was particularly efficient in detecting HBV genomes with multiple large in-frame deletions and spliced variants concomitantly with non-deleted parental genomes. However, an average-12% sequencing error rate per individual reads associated to a low throughput challenged single-nucleotide resolution, polymorphism calling and phasing mutations directly from the sequencing reads. Despite this high error rate, the pairwise identity of MinION HBV consensus genome was consistent with Sanger sequencing method. MinION being under continuous development, further studies are needed to evaluate its potential use for viral infection characterization.
Conflict of interest statement
Figures
Similar articles
-
An Oxford Nanopore Technology-Based Hepatitis B Virus Sequencing Protocol Suitable for Genomic Surveillance Within Clinical Diagnostic Settings.Int J Mol Sci. 2024 Oct 31;25(21):11702. doi: 10.3390/ijms252111702. Int J Mol Sci. 2024. PMID: 39519254 Free PMC article.
-
Single-Molecule Sequencing Reveals Complex Genome Variation of Hepatitis B Virus during 15 Years of Chronic Infection following Liver Transplantation.J Virol. 2016 Jul 27;90(16):7171-7183. doi: 10.1128/JVI.00243-16. Print 2016 Aug 15. J Virol. 2016. PMID: 27252524 Free PMC article.
-
Genome assembly using Nanopore-guided long and error-free DNA reads.BMC Genomics. 2015 Apr 20;16(1):327. doi: 10.1186/s12864-015-1519-z. BMC Genomics. 2015. PMID: 25927464 Free PMC article.
-
Exploring the hepatitis C virus genome using single molecule real-time sequencing.World J Gastroenterol. 2019 Aug 28;25(32):4661-4672. doi: 10.3748/wjg.v25.i32.4661. World J Gastroenterol. 2019. PMID: 31528092 Free PMC article. Review.
-
Recent advances in molecular diagnostics of hepatitis B virus.World J Gastroenterol. 2014 Oct 28;20(40):14615-25. doi: 10.3748/wjg.v20.i40.14615. World J Gastroenterol. 2014. PMID: 25356025 Free PMC article. Review.
Cited by
-
Shotgun metagenome data of a defined mock community using Oxford Nanopore, PacBio and Illumina technologies.Sci Data. 2019 Nov 26;6(1):285. doi: 10.1038/s41597-019-0287-z. Sci Data. 2019. PMID: 31772173 Free PMC article.
-
Viral Metagenomics for Identification of Emerging Viruses in Transfusion Medicine.Viruses. 2022 Nov 4;14(11):2448. doi: 10.3390/v14112448. Viruses. 2022. PMID: 36366546 Free PMC article. Review.
-
Detection of an unusual G8P[8] rotavirus in a Rotarix-vaccinated child with acute gastroenteritis using Nanopore MinION sequencing: A case report.Medicine (Baltimore). 2020 Oct 2;99(40):e22641. doi: 10.1097/MD.0000000000022641. Medicine (Baltimore). 2020. PMID: 33019489 Free PMC article.
-
Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV).Sci Rep. 2019 May 8;9(1):7081. doi: 10.1038/s41598-019-43524-9. Sci Rep. 2019. PMID: 31068626 Free PMC article.
-
Direct sequencing of RNA with MinION Nanopore: detecting mutations based on associations.Nucleic Acids Res. 2019 Dec 16;47(22):e148. doi: 10.1093/nar/gkz907. Nucleic Acids Res. 2019. PMID: 31665473 Free PMC article.
References
-
- Tong S, Revill P. Overview of hepatitis B viral replication and genetic variability. J Hepatol. 2016;64(1 Suppl):S4–S16. doi: 10.1016/j.jhep.2016.01.027 - DOI - PMC - PubMed
-
- Candotti D, Diarra B, Bisseye C, Tao I, Pham Quang K, Sanou M, et al. Molecular characterization of hepatitis B virus in blood donors from Burkina Faso: Prevalence of quasi-subgenotype A3, genotype E, and mixed infections. J Med Virol. 2016;88:2145–2156. doi: 10.1002/jmv.24589 - DOI - PubMed
-
- Sauvage V, Eloit M. Viral metagenomics and blood safety. Transfus Clin Biol. 2016;23:28–38. doi: 10.1016/j.tracli.2015.12.002 - DOI - PMC - PubMed
-
- Rhoads A, Au KF. PacBio Sequencing and its applications. Genomics Proteomics Bioinformatics. 2015;13:278–289. doi: 10.1016/j.gpb.2015.08.002 - DOI - PMC - PubMed
-
- Huang DW, Raley C, Jiang MK, Zheng X, Liang D, Rehman MT, et al. Towards Better Precision Medicine: PacBio Single-Molecule Long Reads Resolve the Interpretation of HIV Drug Resistant Mutation Profiles at Explicit Quasispecies (Haplotype) Level. J Data Mining Genomics Proteomics. 2016;7(1). pii: 182 doi: 10.4172/2153-0602.1000182 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
