

Next-generation sequencing analysis of a cluster of hepatitis C virus infections in a haematology and oncology center
- PMID: 29566084
- PMCID: PMC5864040
- DOI: 10.1371/journal.pone.0194816
Next-generation sequencing analysis of a cluster of hepatitis C virus infections in a haematology and oncology center
Abstract
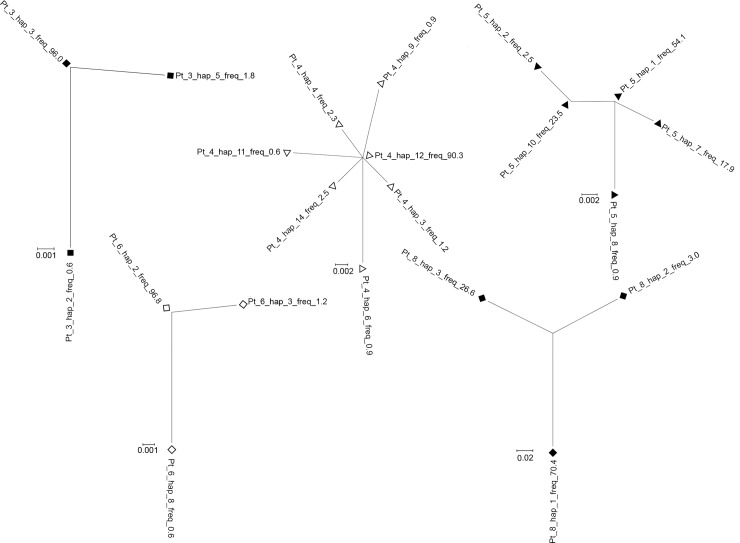
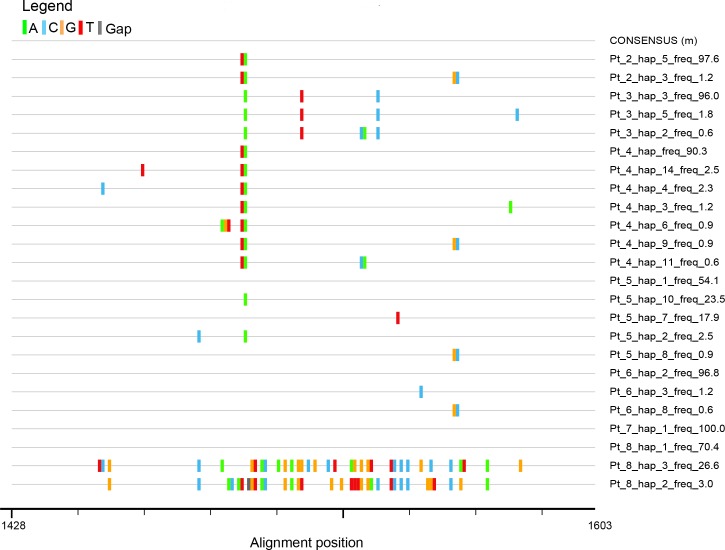
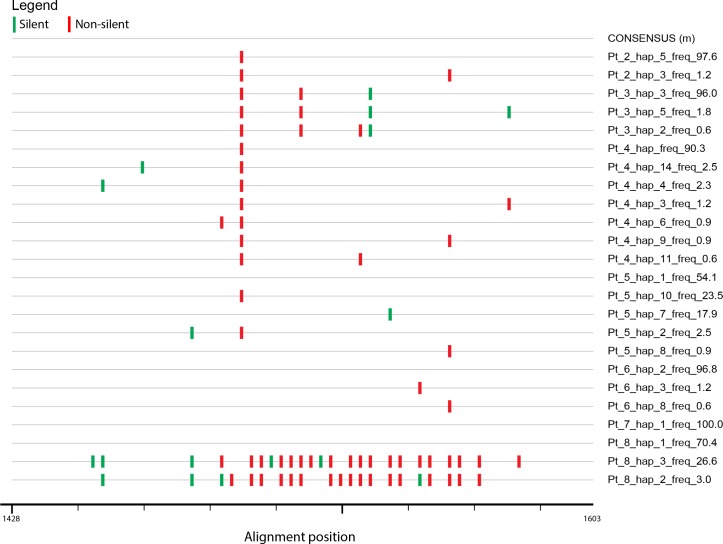
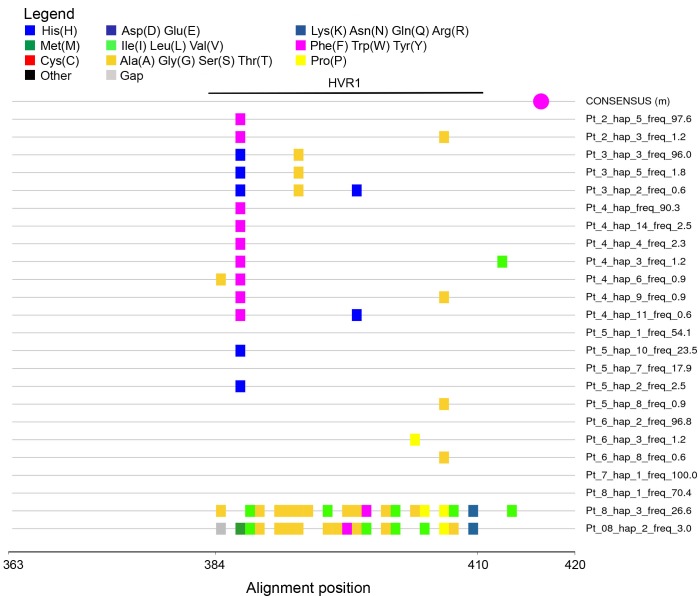
Molecular characterization of early hepatitis C virus (HCV) infection remains rare. Ten out of 78 patients of a hematology/oncology center were found to be HCV RNA positive two to four months after hospitalization. Only two of the ten patients were anti-HCV positive. HCV hypervariable region 1 (HVR1) was amplified in seven patients (including one anti-HCV positive) and analyzed by next generation sequencing (NGS). Genetic variants were reconstructed by Shorah and an empirically established 0.5% variant frequency cut-off was implemented. These sequences were compared by phylogenetic and diversity analyses. Ten unrelated blood donors with newly acquired HCV infection detected at the time of donation (HCV RNA positive and anti-HCV negative) served as controls. One to seven HVR1 variants were found in each patient. Sequences intermixed phylogenetically with no evidence of clustering in individual patients. These sequences were more similar to each other (similarity 95.4% to 100.0%) than to those of controls (similarity 64.8% to 82.6%). An identical predominant variant was present in four patients, whereas other closely related variants dominated in the remaining three patients. In five patients the HCV population was limited to a single variant or one predominant variant and minor variants of less than 10% frequency. In conclusion, NGS analysis of a cluster of HCV infections acquired in the hospital setting revealed the presence of low diversity, very closely related variants in all patients, suggesting an early-stage infection with the same virus. NGS combined with phylogenetic analysis and classical epidemiological analysis could help in tracking of HCV outbreaks.
Conflict of interest statement
Figures

Similar articles
-
Spouse-to-Spouse Transmission and Evolution of Hypervariable Region 1 and 5' Untranslated Region of Hepatitis C Virus Analyzed by Next-Generation Sequencing.PLoS One. 2016 Feb 26;11(2):e0150311. doi: 10.1371/journal.pone.0150311. eCollection 2016. PLoS One. 2016. PMID: 26918636 Free PMC article.
-
An outbreak of HBV and HCV infection in a paediatric oncology ward: epidemiological investigations and prevention of further spread.J Med Virol. 2003 Mar;69(3):331-8. doi: 10.1002/jmv.10293. J Med Virol. 2003. PMID: 12526042
-
Next-generation sequencing studies on the E1-HVR1 region of hepatitis C virus (HCV) from non-high-risk HCV patients living in Punjab and Khyber Pakhtunkhwa, Pakistan.Arch Virol. 2021 Nov;166(11):3049-3059. doi: 10.1007/s00705-021-05203-x. Epub 2021 Aug 27. Arch Virol. 2021. PMID: 34448937
-
Next-generation sequencing for the clinical management of hepatitis C virus infections: does one test fits all purposes?Crit Rev Clin Lab Sci. 2019 Sep;56(6):420-434. doi: 10.1080/10408363.2019.1637394. Epub 2019 Jul 18. Crit Rev Clin Lab Sci. 2019. PMID: 31317801 Review.
-
Next-Generation Sequencing Methods for Near-Real-Time Molecular Epidemiology of HIV and HCV.Rev Med Virol. 2024 Nov;34(6):e70001. doi: 10.1002/rmv.70001. Rev Med Virol. 2024. PMID: 39428551 Review.
Cited by
-
Hepacivirus A Infection in Horses Defines Distinct Envelope Hypervariable Regions and Elucidates Potential Roles of Viral Strain and Adaptive Immune Status in Determining Envelope Diversity and Infection Outcome.J Virol. 2018 Aug 29;92(18):e00314-18. doi: 10.1128/JVI.00314-18. Print 2018 Sep 15. J Virol. 2018. Retraction in: J Virol. 2020 Dec 9;95(1):e01963-20. doi: 10.1128/JVI.01963-20. PMID: 29976666 Free PMC article. Retracted.
-
The emergence of new trends in clinical laboratory diagnosis.Saudi Med J. 2020 Nov;41(11):1175-1180. doi: 10.15537/smj.2020.11.25455. Saudi Med J. 2020. PMID: 33130836 Free PMC article. Review.
-
Molecular and spatial epidemiology of HCV among people who inject drugs in Boston, Massachusetts.PLoS One. 2022 Aug 25;17(8):e0266216. doi: 10.1371/journal.pone.0266216. eCollection 2022. PLoS One. 2022. PMID: 36006966 Free PMC article.
References
-
- Farci P, Shimoda A, Coiana A, Diaz G, Peddis G, Melpolder JC, et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 2000;288(5464):339–44. . - PubMed
-
- Domingo E, Sheldon J, Perales C. Viral quasispecies evolution. Microbiology and molecular biology reviews: MMBR. 2012;76(2):159–216. doi: 10.1128/MMBR.05023-11 ; PubMed Central PMCID: PMC3372249. - DOI - PMC - PubMed
-
- Wang GP, Sherrill-Mix SA, Chang KM, Quince C, Bushman FD. Hepatitis C virus transmission bottlenecks analyzed by deep sequencing. J Virol. 2010;84(12):6218–28. doi: 10.1128/JVI.02271-09 ; PubMed Central PMCID: PMCPMC2876626. - DOI - PMC - PubMed
-
- Bull RA, Luciani F, McElroy K, Gaudieri S, Pham ST, Chopra A, et al. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS pathogens. 2011;7(9):e1002243 doi: 10.1371/journal.ppat.1002243 ; PubMed Central PMCID: PMCPMC3164670. - DOI - PMC - PubMed
-
- Li H, Stoddard MB, Wang S, Blair LM, Giorgi EE, Parrish EH, et al. Elucidation of hepatitis C virus transmission and early diversification by single genome sequencing. PLoS pathogens. 2012;8(8):e1002880 doi: 10.1371/journal.ppat.1002880 ; PubMed Central PMCID: PMCPMC3426529. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
