

The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression
- PMID: 29567829
- PMCID: PMC6225783
- DOI: 10.1158/2159-8290.CD-17-1134
The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression
Erratum in
-
Correction: The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression.Cancer Discov. 2020 Dec;10(12):1988. doi: 10.1158/2159-8290.CD-20-1573. Cancer Discov. 2020. PMID: 33262184 No abstract available.
Abstract
We found that the cancerous pancreas harbors a markedly more abundant microbiome compared with normal pancreas in both mice and humans, and select bacteria are differentially increased in the tumorous pancreas compared with gut. Ablation of the microbiome protects against preinvasive and invasive pancreatic ductal adenocarcinoma (PDA), whereas transfer of bacteria from PDA-bearing hosts, but not controls, reverses tumor protection. Bacterial ablation was associated with immunogenic reprogramming of the PDA tumor microenvironment, including a reduction in myeloid-derived suppressor cells and an increase in M1 macrophage differentiation, promoting TH1 differentiation of CD4+ T cells and CD8+ T-cell activation. Bacterial ablation also enabled efficacy for checkpoint-targeted immunotherapy by upregulating PD-1 expression. Mechanistically, the PDA microbiome generated a tolerogenic immune program by differentially activating select Toll-like receptors in monocytic cells. These data suggest that endogenous microbiota promote the crippling immune-suppression characteristic of PDA and that the microbiome has potential as a therapeutic target in the modulation of disease progression.Significance: We found that a distinct and abundant microbiome drives suppressive monocytic cellular differentiation in pancreatic cancer via selective Toll-like receptor ligation leading to T-cell anergy. Targeting the microbiome protects against oncogenesis, reverses intratumoral immune tolerance, and enables efficacy for checkpoint-based immunotherapy. These data have implications for understanding immune suppression in pancreatic cancer and its reversal in the clinic. Cancer Discov; 8(4); 403-16. ©2018 AACR.See related commentary by Riquelme et al., p. 386This article is highlighted in the In This Issue feature, p. 371.
©2018 American Association for Cancer Research.
Conflict of interest statement
Competing financial interests
None of the authors have any competing financial interests to declare.
The authors declare no potential conflicts of interest.
Figures
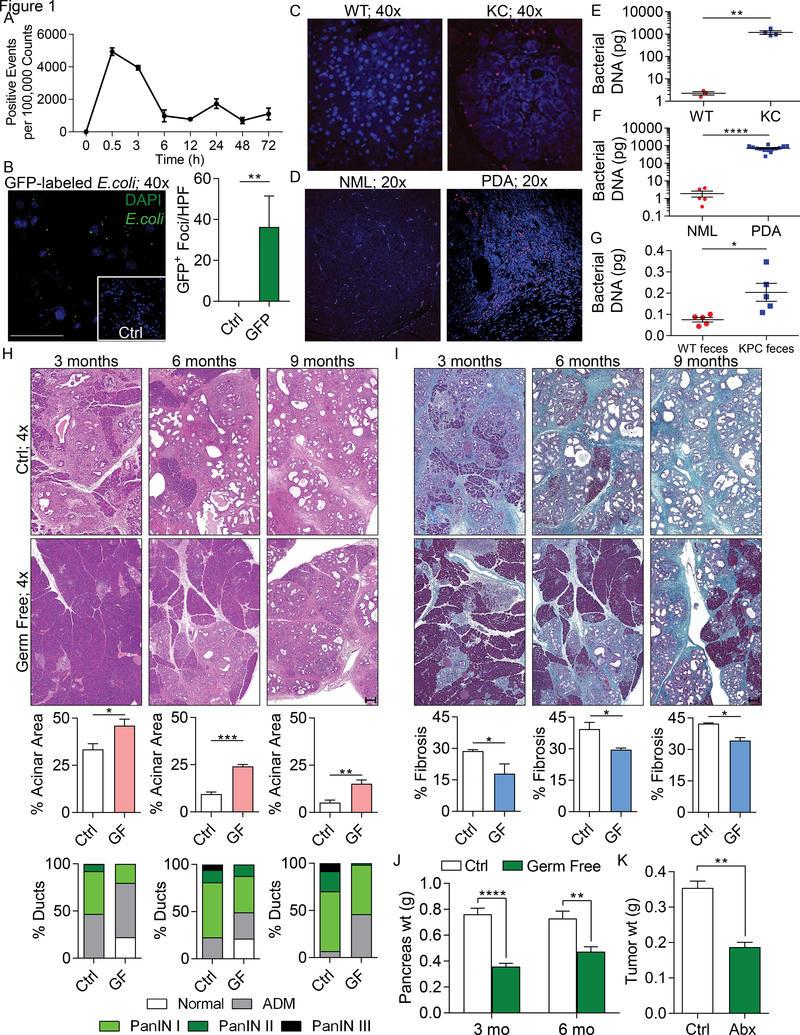
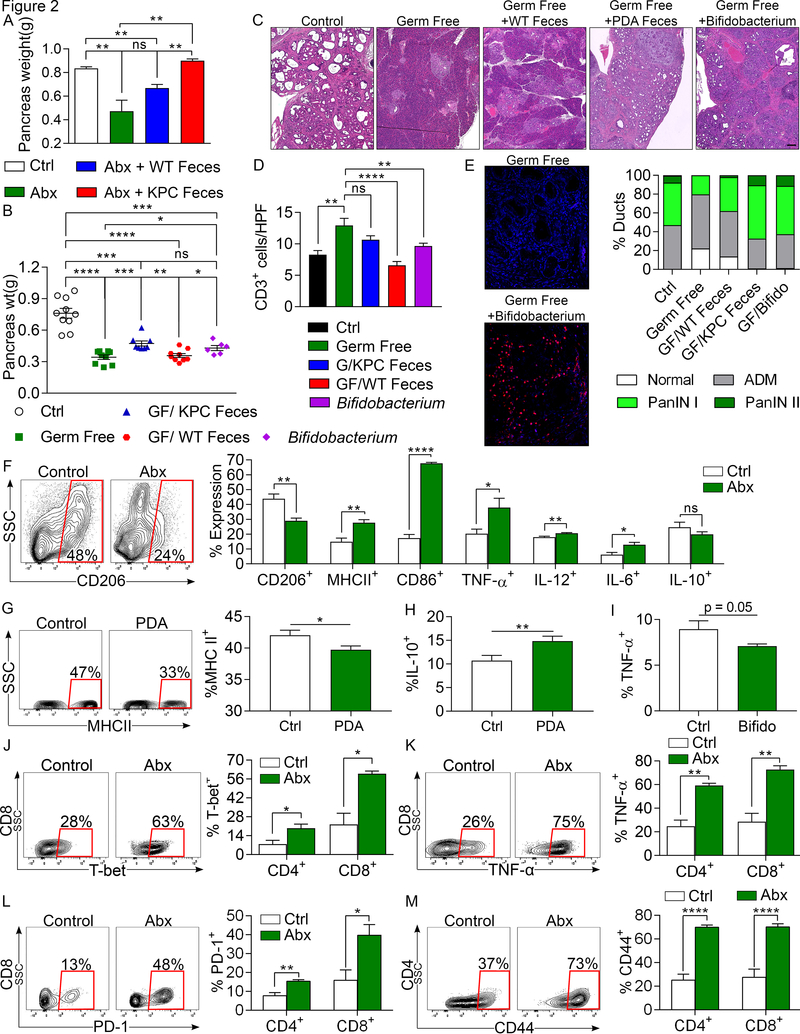
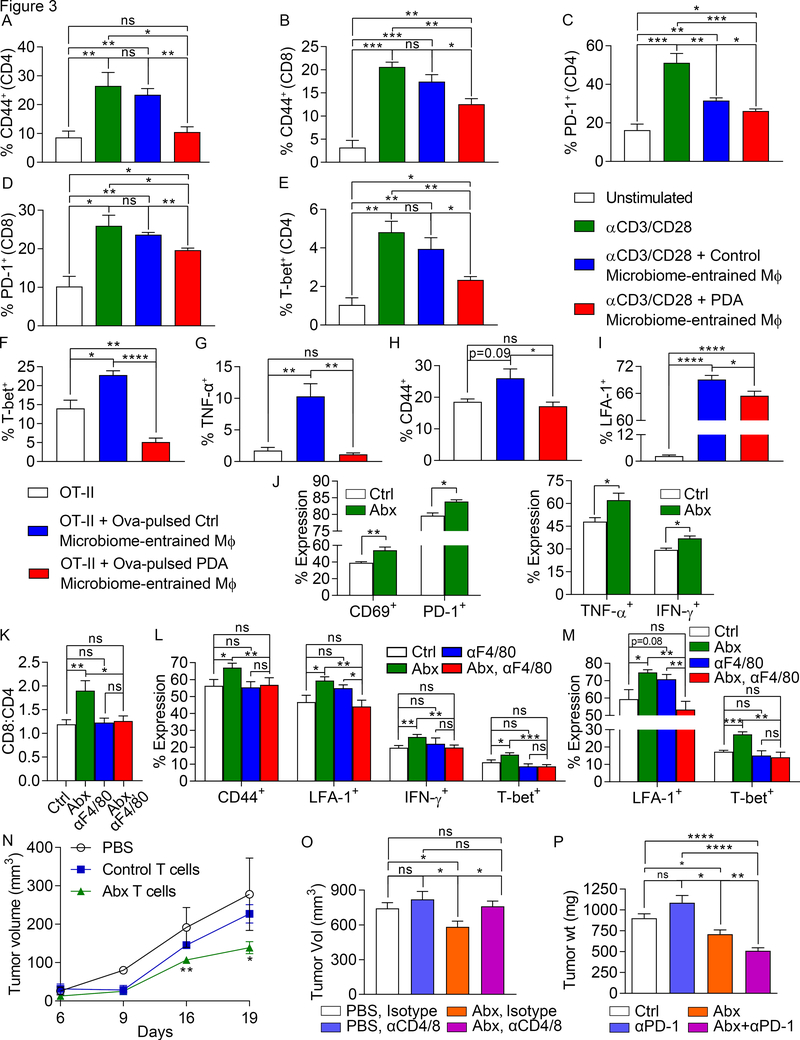
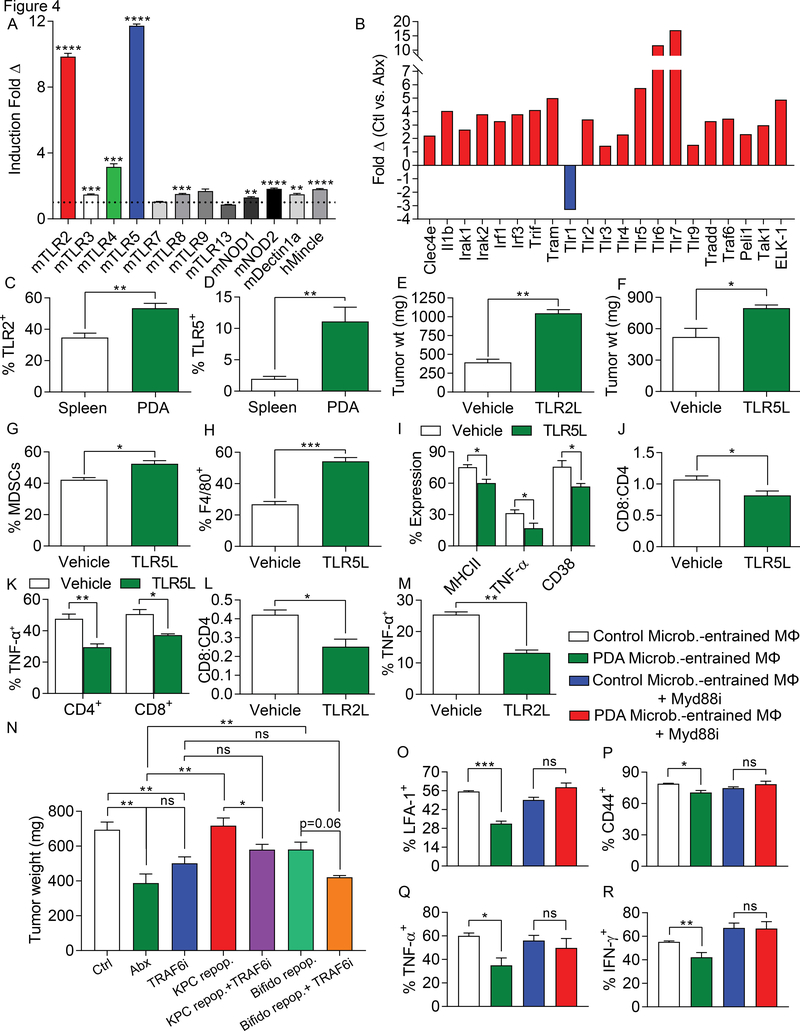
Comment in
-
Immunotherapy for Pancreatic Cancer: More Than Just a Gut Feeling.Cancer Discov. 2018 Apr;8(4):386-388. doi: 10.1158/2159-8290.CD-18-0123. Cancer Discov. 2018. PMID: 29610286
-
Microbiome promotes pancreatic cancer.Nat Rev Gastroenterol Hepatol. 2018 Jun;15(6):328. doi: 10.1038/s41575-018-0013-x. Nat Rev Gastroenterol Hepatol. 2018. PMID: 29674642 No abstract available.
-
Microbial marauders: pancreatic microbiota and its impact on carcinogenesis.Ann Transl Med. 2018 Nov;6(Suppl 1):S63. doi: 10.21037/atm.2018.10.34. Ann Transl Med. 2018. PMID: 30613638 Free PMC article. No abstract available.
References
-
- Siegel RL, Miller KD and Jemal A (2016) Cancer statistics, 2016. CA: a cancer journal for clinicians, 66, 7–30. - PubMed
-
- Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, Zambirinis CP, Fallon NC, Rehman A, Pylayeva-Gupta Y et al. (2012) MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. The Journal of experimental medicine, 209, 1671–1687. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
