

BEARscc determines robustness of single-cell clusters using simulated technical replicates
- PMID: 29567991
- PMCID: PMC5864873
- DOI: 10.1038/s41467-018-03608-y
BEARscc determines robustness of single-cell clusters using simulated technical replicates
Abstract
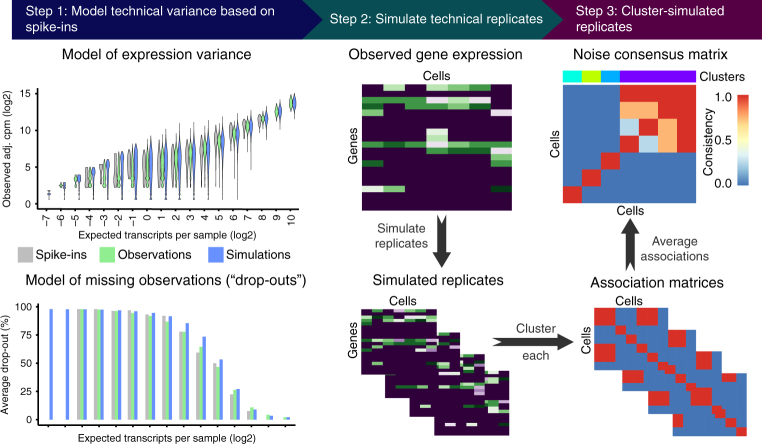
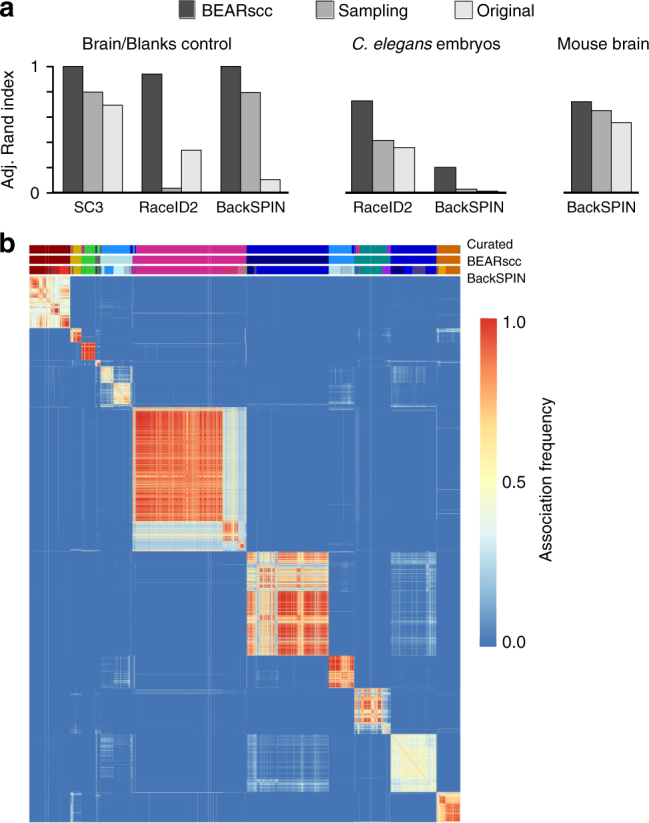
Single-cell messenger RNA sequencing (scRNA-seq) has emerged as a powerful tool to study cellular heterogeneity within complex tissues. Subpopulations of cells with common gene expression profiles can be identified by applying unsupervised clustering algorithms. However, technical variance is a major confounding factor in scRNA-seq, not least because it is not possible to replicate measurements on the same cell. Here, we present BEARscc, a tool that uses RNA spike-in controls to simulate experiment-specific technical replicates. BEARscc works with a wide range of existing clustering algorithms to assess the robustness of clusters to technical variation. We demonstrate that the tool improves the unsupervised classification of cells and facilitates the biological interpretation of single-cell RNA-seq experiments.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
