

Identification of p38 MAPK as a novel therapeutic target for Friedreich's ataxia
- PMID: 29568068
- PMCID: PMC5864720
- DOI: 10.1038/s41598-018-23168-x
Identification of p38 MAPK as a novel therapeutic target for Friedreich's ataxia
Abstract
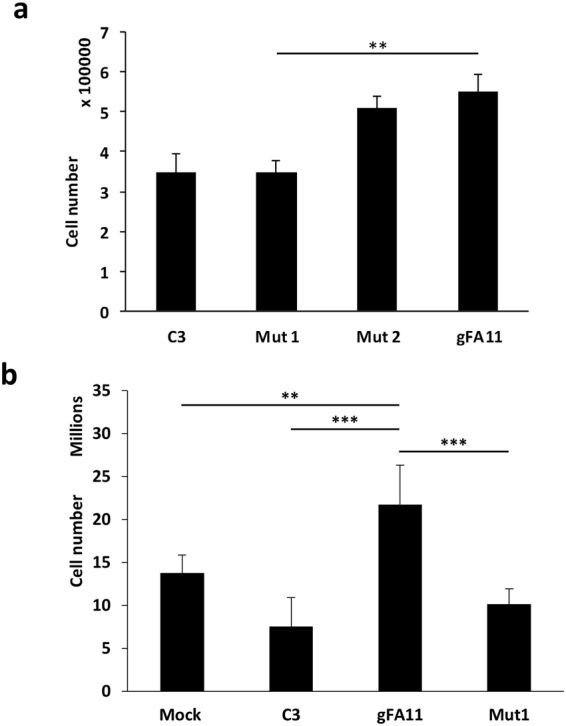
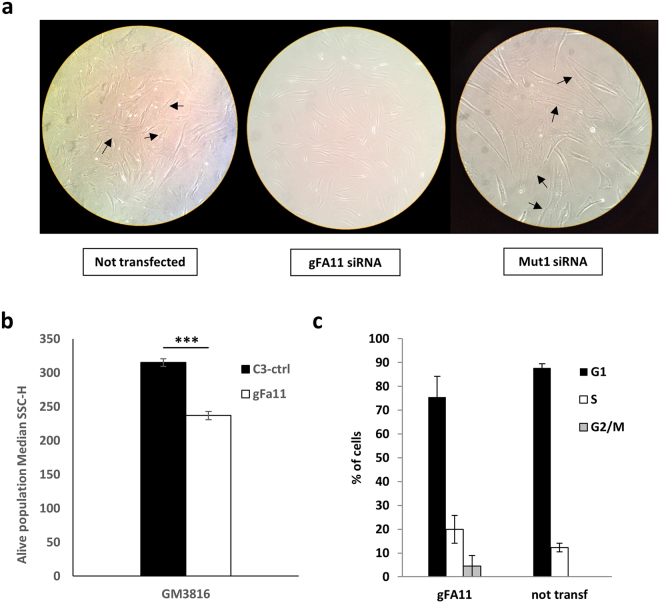
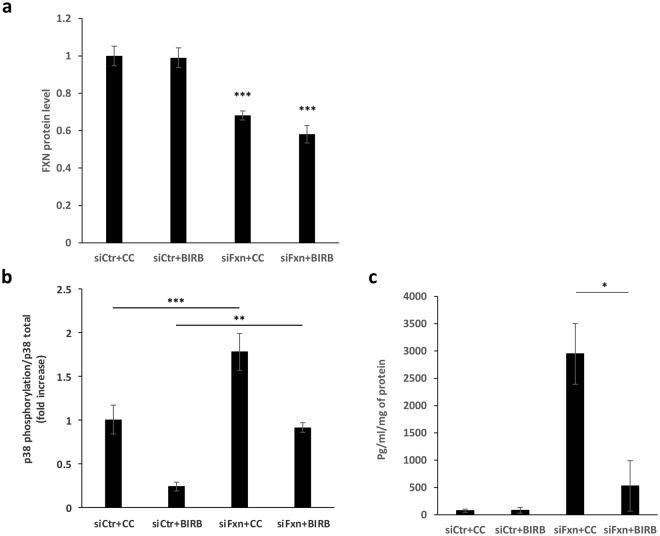
Friedreich ataxia (FRDA) is an autosomal recessive neuro- and cardio-degenerative disorder caused by decreased expression of frataxin, a protein that localizes to mitochondria and is critical for iron-sulfur-cluster (ISC) assembly. There are no proven effective treatments for FRDA. We previously screened a random shRNA library and identified a synthetic shRNA (gFA11) that reverses the growth defect of FRDA cells in culture. We now report that gFA11 decreases cytokine secretion in primary FRDA fibroblasts and reverts other changes associated with cell senescence. The gene-expression profile induced by gFA11 is remarkably similar to the gene-expression profile induced by the p38 MAPK inhibitor SB203580. We found that p38 phosphorylation, indicating activation of the p38 pathway, is higher in FRDA cells than in normal control cells, and that siRNA knockdown of frataxin in normal fibroblasts also increases p38 phosphorylation. Treatment of FRDA cells with p38 inhibitors recapitulates the reversal of the slow-growth phenotype induced by clone gFA11. These data highlight the involvement of the p38 MAPK pathway in the pathogenesis of FRDA and the potential use of p38 inhibitors as a treatment for FRDA.
Conflict of interest statement
Robert B. Wilson and Yongping Wang have patents, in conjunction with the University of Pennsylvania, related to the construction and methods of use of random shRNA-expressing libraries. M. Grazia Cotticelli, Shujuan Xia, Avinash Kaur, Yongping Wang, John W. Tobias, and Robert B. Wilson have a provisional patent application, in conjunction with the University of Pennsylvania, on the use of p38 inhibitors for the treatment of FRDA; this provisional patent was licensed to FA Therapeutics LLC, in which Robert B. Wilson has a 5% equity interest.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
