

Cigarette smoke enhances oncogene addiction to c-MET and desensitizes EGFR-expressing non-small cell lung cancer to EGFR TKIs
- PMID: 29570930
- PMCID: PMC5928373
- DOI: 10.1002/1878-0261.12193
Cigarette smoke enhances oncogene addiction to c-MET and desensitizes EGFR-expressing non-small cell lung cancer to EGFR TKIs
Abstract
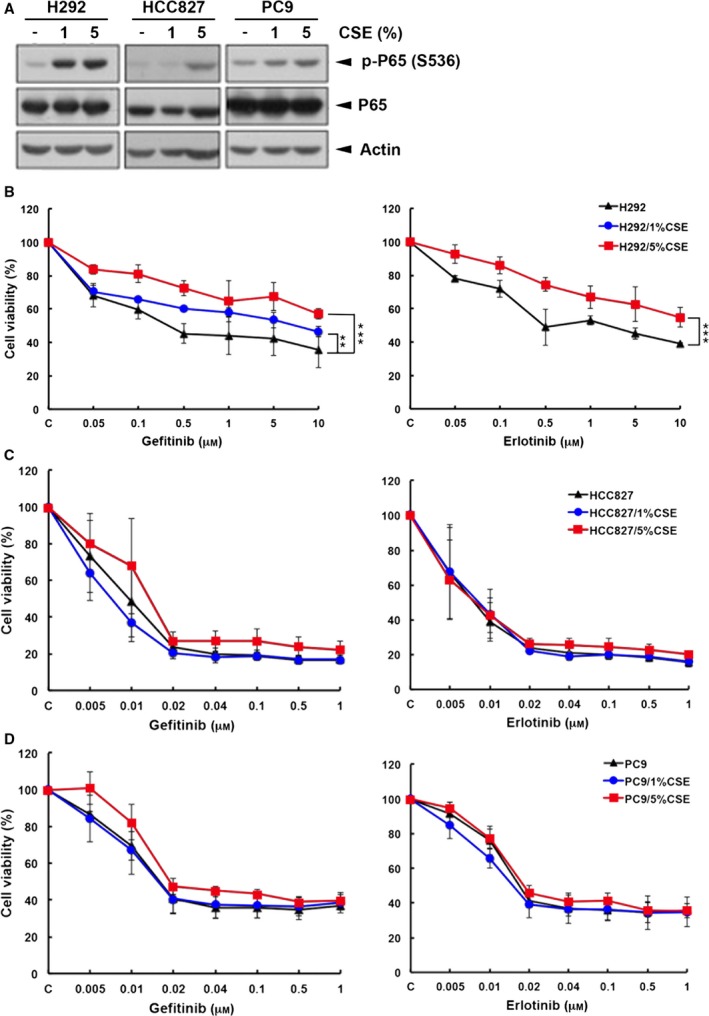
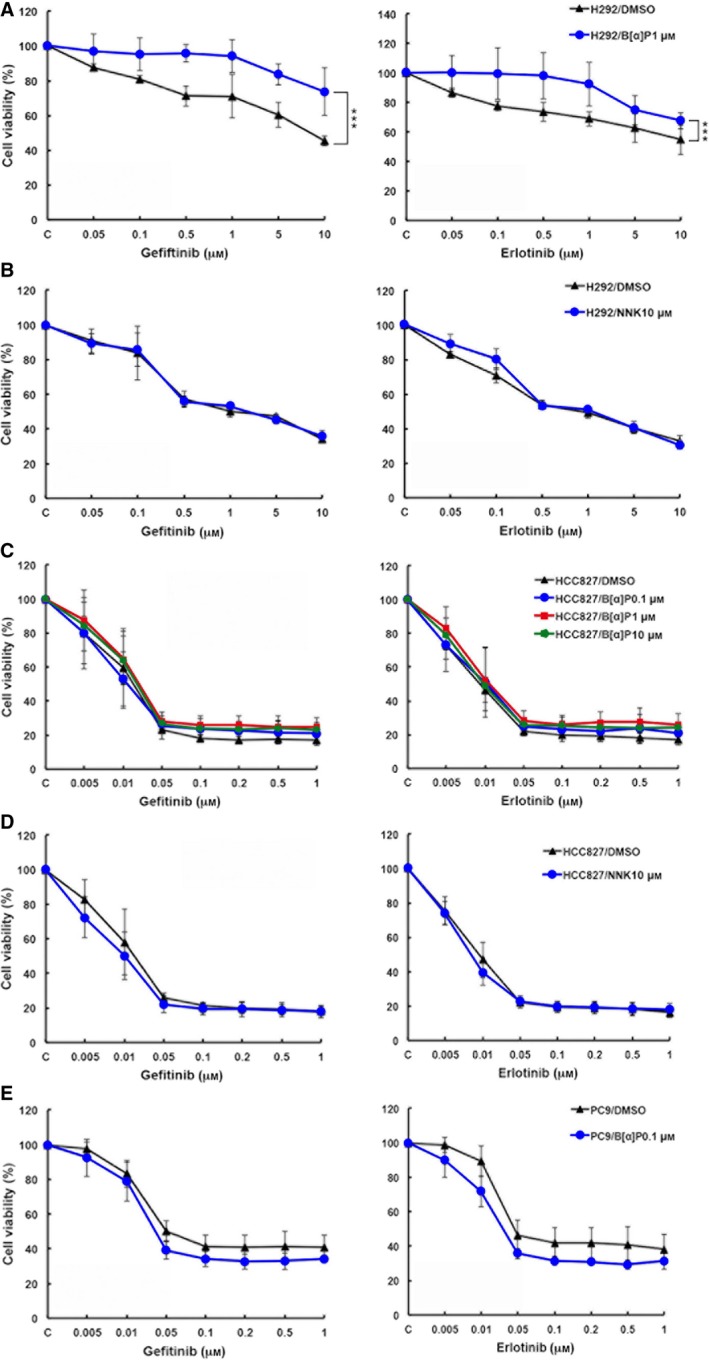
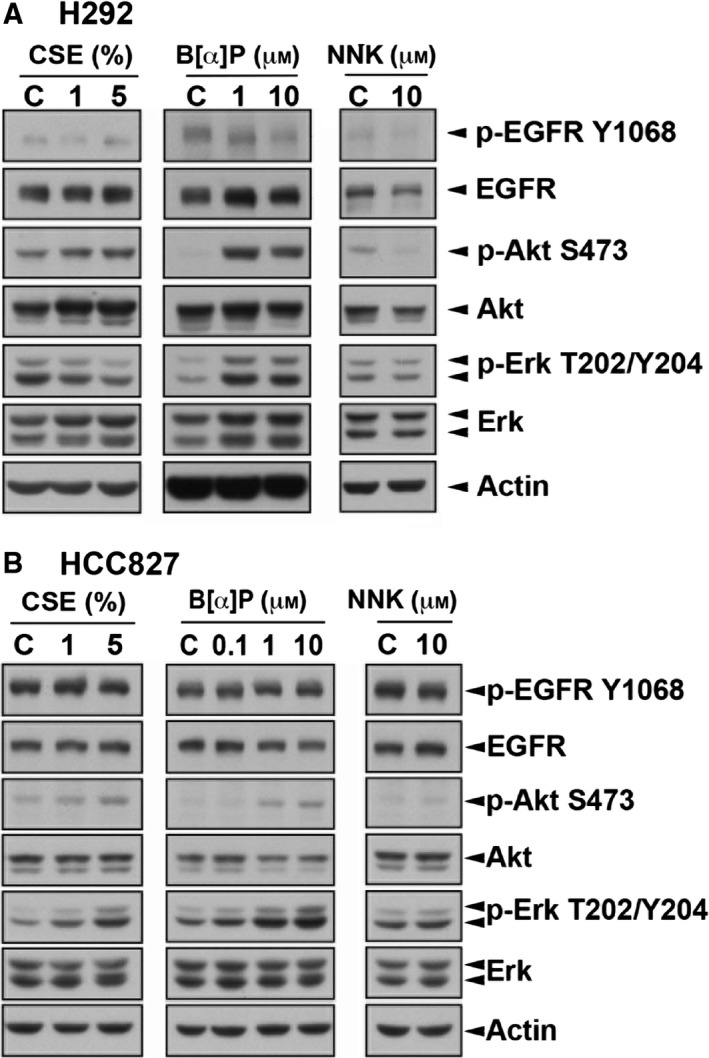
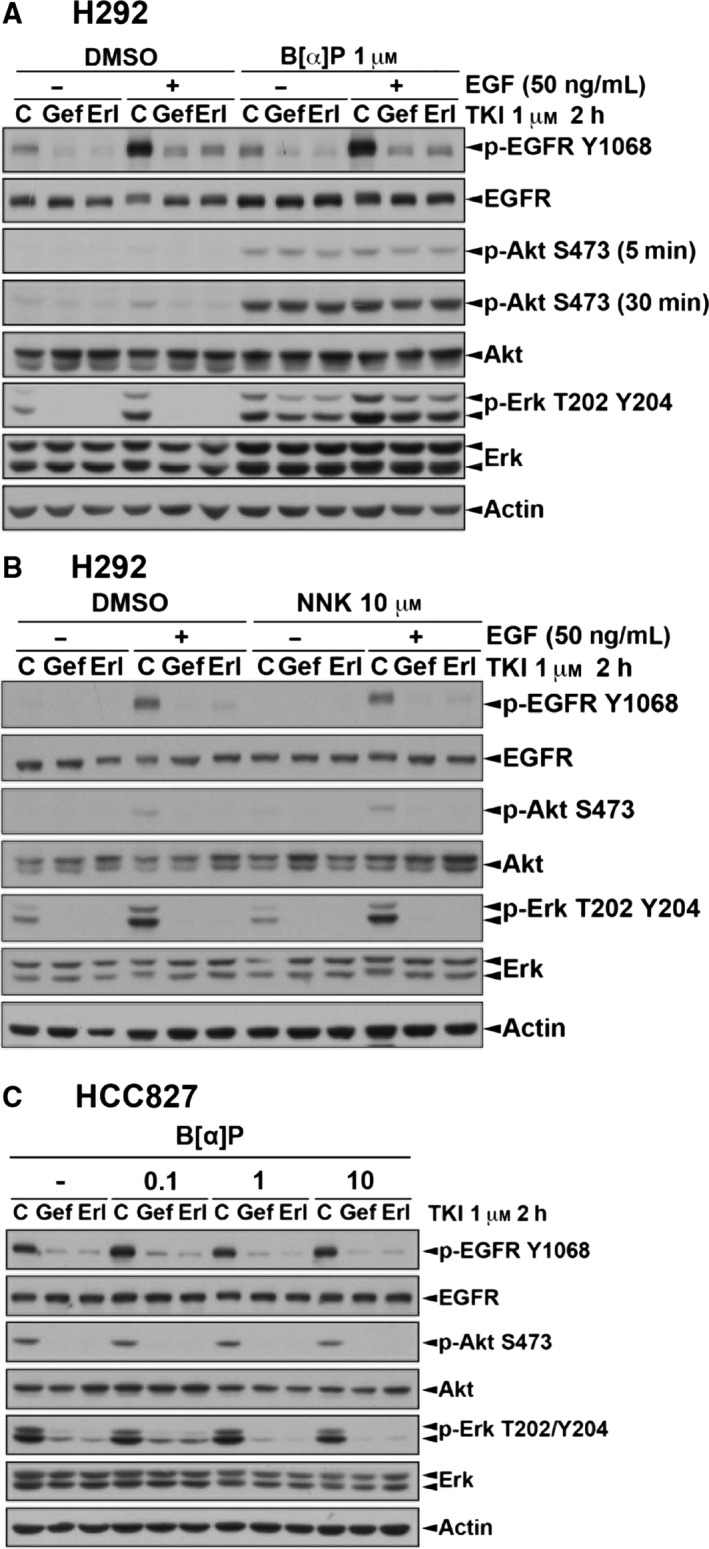
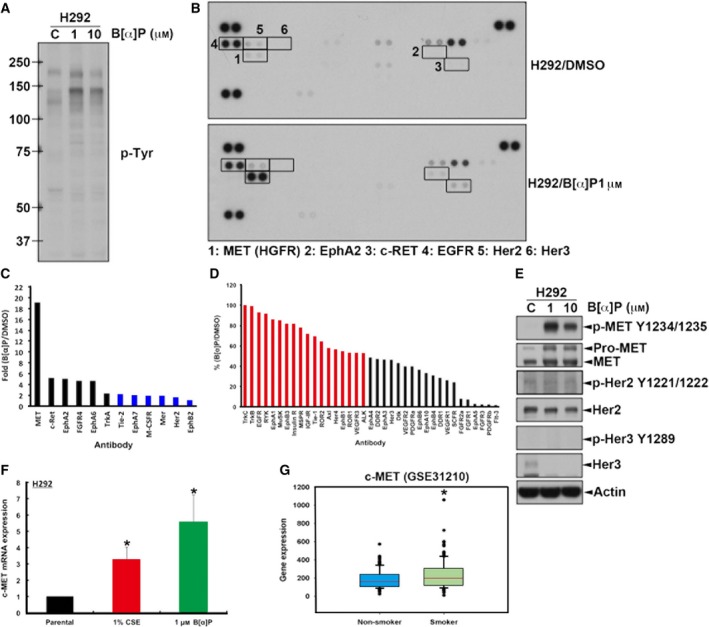
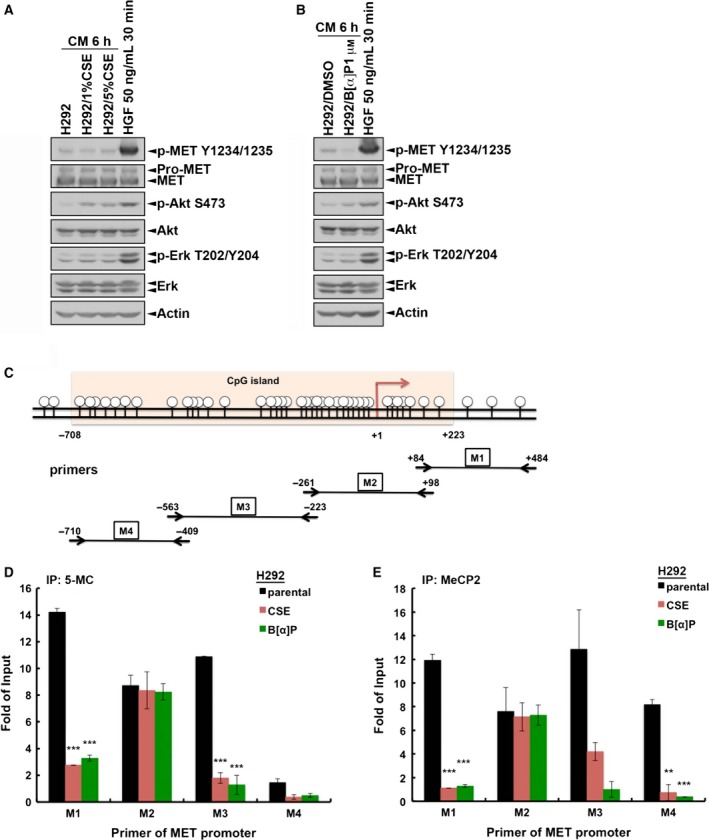
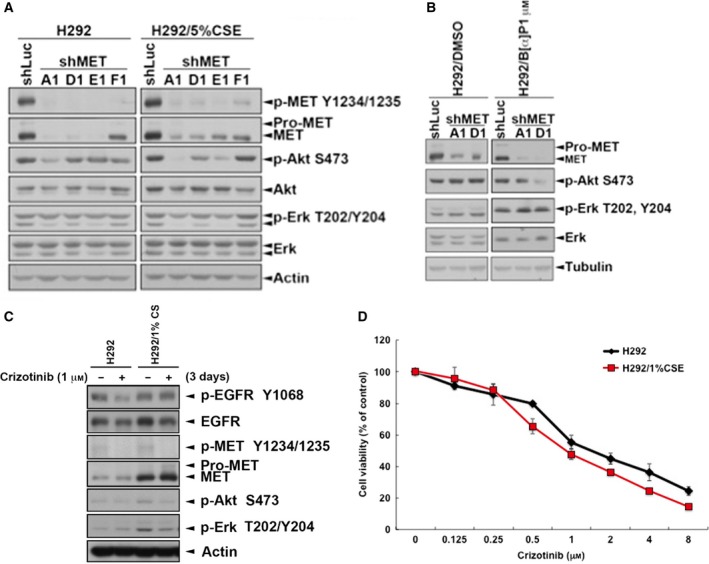
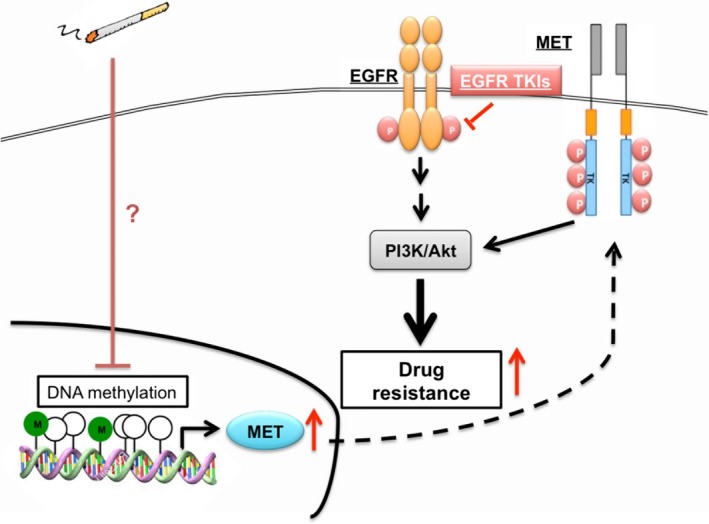
Cigarette smoking is one of the leading risks for lung cancer and is associated with the insensitivity of non-small cell lung cancer (NSCLC) to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). However, it remains undetermined whether and how cigarette smoke affects the therapeutic efficacy of EGFR TKIs. In this study, our data showed that chronic exposure to cigarette smoke extract (CSE) or tobacco smoke-derived carcinogen benzo[α]pyrene, B[α]P, but not nicotine-derived nitrosamine ketone (NNK), reduced the sensitivity of wild-type EGFR-expressing NSCLC cells to EGFR TKIs. Treatment with TKIs almost abolished EGFR tyrosine kinase activity but did not show an inhibitory effect on downstream Akt and ERK pathways in B[α]P-treated NSCLC cells. CSE and B[α]P transcriptionally upregulate c-MET and activate its downstream Akt pathway, which is not inhibited by EGFR TKIs. Silencing of c-MET reduces B[α]P-induced Akt activation. The CSE-treated NSCLC cells are sensitive to the c-MET inhibitor crizotinib. These findings suggest that cigarette smoke augments oncogene addiction to c-MET in NSCLC cells and that MET inhibitors may show clinical benefits for lung cancer patients with a smoking history.
Keywords: EGFR-TKI; benzo[α]pyrene; c-MET; cigarette smoke; lung cancer.
© 2018 The Authors. Published by FEBS Press and John Wiley & Sons Ltd.
Figures
Similar articles
-
Src mediates cigarette smoke-induced resistance to tyrosine kinase inhibitors in NSCLC cells.Mol Cancer Ther. 2013 Aug;12(8):1579-90. doi: 10.1158/1535-7163.MCT-12-1029. Epub 2013 May 17. Mol Cancer Ther. 2013. PMID: 23686837 Free PMC article.
-
Leukocyte Cell-Derived Chemotaxin 2 Retards Non-Small Cell Lung Cancer Progression Through Antagonizing MET and EGFR Activities.Cell Physiol Biochem. 2018;51(1):337-355. doi: 10.1159/000495233. Epub 2018 Nov 19. Cell Physiol Biochem. 2018. PMID: 30453282
-
Cigarette smoke extract exposure induces EGFR-TKI resistance in EGFR-mutated NSCLC via mediating Src activation and EMT.Lung Cancer. 2016 Mar;93:35-42. doi: 10.1016/j.lungcan.2015.12.007. Epub 2015 Dec 30. Lung Cancer. 2016. PMID: 26898612
-
Management and future directions in non-small cell lung cancer with known activating mutations.Am Soc Clin Oncol Educ Book. 2014:e353-65. doi: 10.14694/EdBook_AM.2014.34.e353. Am Soc Clin Oncol Educ Book. 2014. PMID: 24857124 Review.
-
Does c-Met remain a rational target for therapy in patients with EGFR TKI-resistant non-small cell lung cancer?Cancer Treat Rev. 2017 Dec;61:70-81. doi: 10.1016/j.ctrv.2017.10.003. Epub 2017 Oct 25. Cancer Treat Rev. 2017. PMID: 29121501 Review.
Cited by
-
EGFR-AS1/HIF2A regulates the expression of FOXP3 to impact the cancer stemness of smoking-related non-small cell lung cancer.Ther Adv Med Oncol. 2019 Jun 13;11:1758835919855228. doi: 10.1177/1758835919855228. eCollection 2019. Ther Adv Med Oncol. 2019. PMID: 31275431 Free PMC article.
-
Cigarette smoke-induced LKB1/AMPK pathway deficiency reduces EGFR TKI sensitivity in NSCLC.Oncogene. 2021 Feb;40(6):1162-1175. doi: 10.1038/s41388-020-01597-1. Epub 2020 Dec 17. Oncogene. 2021. PMID: 33335306 Free PMC article.
-
Tobacco Smoking, Lung Cancer, and Therapy in Iraq: Current Perspective.Front Public Health. 2018 Oct 26;6:311. doi: 10.3389/fpubh.2018.00311. eCollection 2018. Front Public Health. 2018. PMID: 30416993 Free PMC article.
-
The Development and Role of Capmatinib in the Treatment of MET-Dysregulated Non-Small Cell Lung Cancer-A Narrative Review.Cancers (Basel). 2023 Jul 10;15(14):3561. doi: 10.3390/cancers15143561. Cancers (Basel). 2023. PMID: 37509224 Free PMC article. Review.
-
Oncogene addiction and tumor mutational burden in non-small-cell lung cancer: Clinical significance and limitations.Thorac Cancer. 2020 Feb;11(2):205-215. doi: 10.1111/1759-7714.13246. Epub 2019 Dec 4. Thorac Cancer. 2020. PMID: 31799812 Free PMC article. Review.
References
-
- Adams JD, O'Mara‐Adams KJ and Hoffmann D (1987) Toxic and carcinogenic agents in undiluted mainstream smoke and sidestream smoke of different types of cigarettes. Carcinogenesis 8, 729–731. - PubMed
-
- Askari MD, Tsao MS and Schuller HM (2005) The tobacco‐specific carcinogen, 4‐ (methylnitrosamino)‐1‐ (3‐pyridyl)‐1‐butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta‐adrenergic transactivation of EGF receptors. J Cancer Res Clin Oncol 131, 639–648. - PubMed
-
- Beau‐Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, Legrain M, Mennecier B, Wihlm JM, Massard G et al (2008) MET gene copy number in non‐small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol 3, 331–339. - PubMed
-
- Bell DW, Lynch TJ, Haserlat SM, Harris PL, Okimoto RA, Brannigan BW, Sgroi DC, Muir B, Riemenschneider MJ, Iacona RB et al (2005) Epidermal growth factor receptor mutations and gene amplification in non‐small‐cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol 23, 8081–8092. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
