

Compound heterozygosity for loss-of-function FARSB variants in a patient with classic features of recessive aminoacyl-tRNA synthetase-related disease
- PMID: 29573043
- PMCID: PMC5992071
- DOI: 10.1002/humu.23424
Compound heterozygosity for loss-of-function FARSB variants in a patient with classic features of recessive aminoacyl-tRNA synthetase-related disease
Abstract
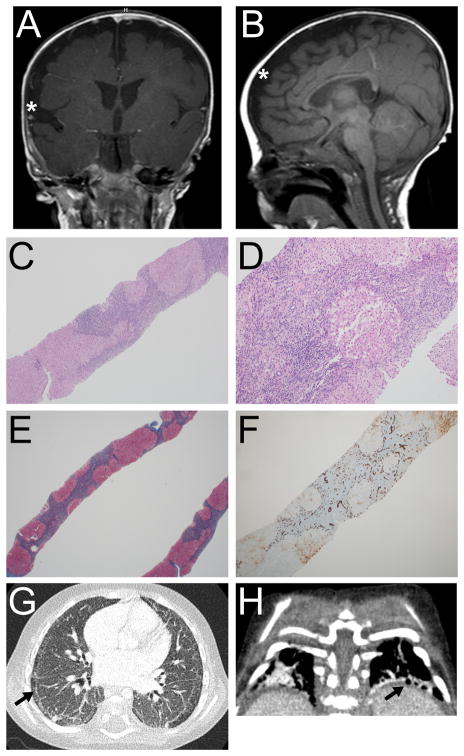
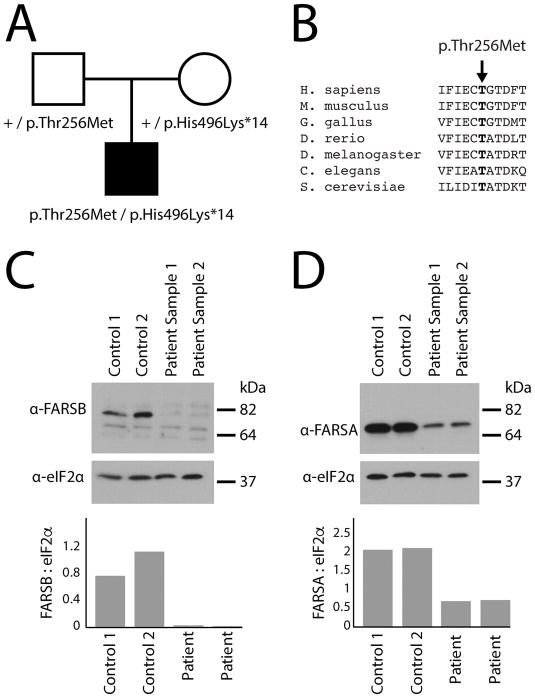
Aminoacyl-tRNA synthetases (ARSs) are ubiquitously expressed enzymes that ligate amino acids onto tRNA molecules. Genes encoding ARSs have been implicated in phenotypically diverse dominant and recessive human diseases. The charging of tRNAPHE with phenylalanine is performed by a tetrameric enzyme that contains two alpha (FARSA) and two beta (FARSB) subunits. To date, mutations in the genes encoding these subunits (FARSA and FARSB) have not been implicated in any human disease. Here, we describe a patient with a severe, lethal, multisystem, developmental phenotype who was compound heterozygous for FARSB variants: p.Thr256Met and p.His496Lysfs*14. Expression studies using fibroblasts isolated from the proband revealed a severe depletion of both FARSB and FARSA protein levels. These data indicate that the FARSB variants destabilize total phenylalanyl-tRNA synthetase levels, thus causing a loss-of-function effect. Importantly, our patient shows strong phenotypic overlap with patients that have recessive diseases associated with other ARS loci; these observations strongly support the pathogenicity of the identified FARSB variants and are consistent with the essential function of phenylalanyl-tRNA synthetase in human cells. In sum, our clinical, genetic, and functional analyses revealed the first FARSB variants associated with a human disease phenotype and expand the locus heterogeneity of ARS-related human disease.
Keywords: FARSB; aminoacyl-tRNA synthetase; developmental syndrome; loss-of-function mutations; phenylalanyl-tRNA synthetase; recessive disease.
© 2018 Wiley Periodicals, Inc.
Conflict of interest statement
The authors have no conflicts to declare.
Figures
References
-
- Almalki A, Alston CL, Parker A, Simonic I, Mehta SG, He L, Reza M, Oliveira JMA, Lightowlers RN, McFarland R, Taylor RW, Chrzanowska-Lightowlers ZMA. Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. Biochim Biophys Acta. 2014;1842:56–64. - PMC - PubMed
-
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin S-Q, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. 2003;72:1293–1299. - PMC - PubMed
-
- Antonellis A, Green ED. The role of aminoacyl-tRNA synthetases in genetic diseases. Annual review of genomics and human genetics. 2008;9:87–107. - PubMed
-
- Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. 2006;62:1125–1132. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
