

To die or not to die: death signaling in nonalcoholic fatty liver disease
- PMID: 29574534
- PMCID: PMC6061666
- DOI: 10.1007/s00535-018-1451-5
To die or not to die: death signaling in nonalcoholic fatty liver disease
Abstract
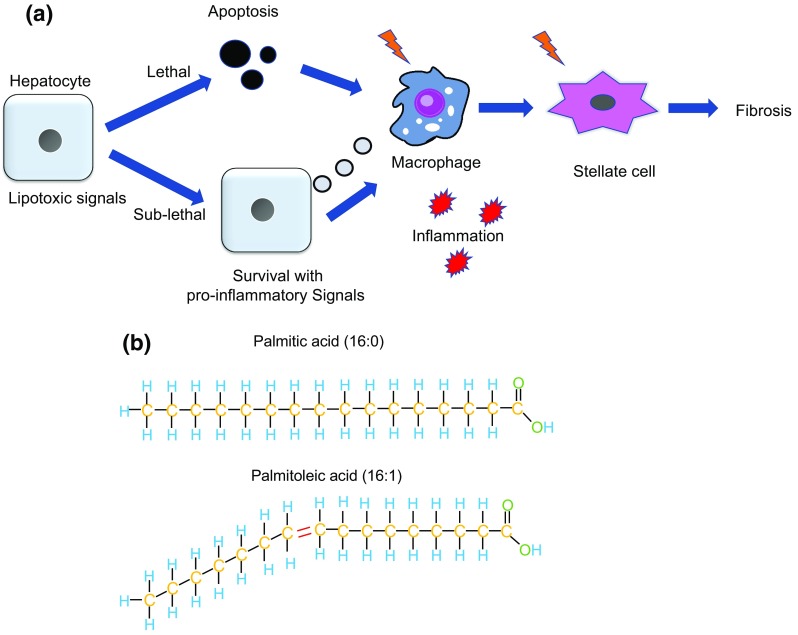
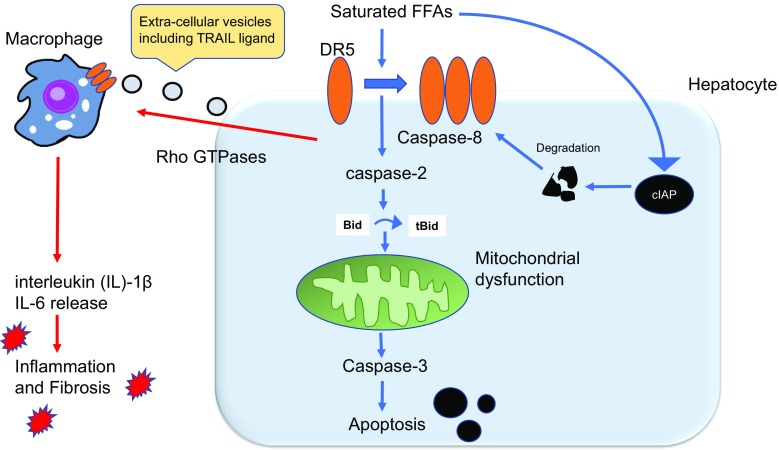
Non-alcoholic fatty liver disease (NAFLD) is an emerging liver disease worldwide. In subset of patients, NAFLD progresses to its advanced form, nonalcoholic steatohepatitis (NASH), which is accompanied with inflammation and fibrosis. Saturated free fatty acid-induced hepatocyte apoptosis is a feature of NASH. Death signaling in NASH does not always result in apoptosis, but can alternatively lead to the survival of cells presenting signs of pro-inflammatory and pro-fibrotic signals. With the current lack of established treatments for NASH, it is important to understand the molecular mechanisms responsible for disease development and progression. This review focuses on the latest findings in hepatocyte death signaling and discusses possible targets for intervention, including caspases, death receptor and c-Jun N-terminal kinase 1 signaling, oxidative stress, and endoplasmic reticulum stress, as well as epigenomic factors.
Keywords: Apoptosis; Endoplasmic reticulum stress; Free fatty acids; Non-alcoholic fatty liver disease; Nonalcoholic steatohepatitis.
Figures



References
-
- Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140(1):124–131. - PubMed
-
- Seki Y, Kakizaki S, Horiguchi N, et al. Prevalence of nonalcoholic steatohepatitis in Japanese patients with morbid obesity undergoing bariatric surgery. J Gastroenterol. 2016;51(3):281–289. - PubMed
-
- Watanabe S, Hashimoto E, Ikejima K, et al. Evidence-based clinical practice guidelines for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. J Gastroenterol. 2015;50(4):364–377. - PubMed
-
- Yan J, Xie W, Ou WN, et al. Epidemiological survey and risk factor analysis of fatty liver disease of adult residents, Beijing, China. J Gastroenterol Hepatol. 2013;28(10):1654–1659. - PubMed
-
- Tokushige K, Hashimoto E, Kodama K. Hepatocarcinogenesis in non-alcoholic fatty liver disease in Japan. J Gastroenterol Hepatol. 2013;28(Suppl 4):88–92. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
