

Elucidating the diagnostic odyssey of 22q11.2 deletion syndrome
- PMID: 29575622
- PMCID: PMC5873609
- DOI: 10.1002/ajmg.a.38645
Elucidating the diagnostic odyssey of 22q11.2 deletion syndrome
Abstract
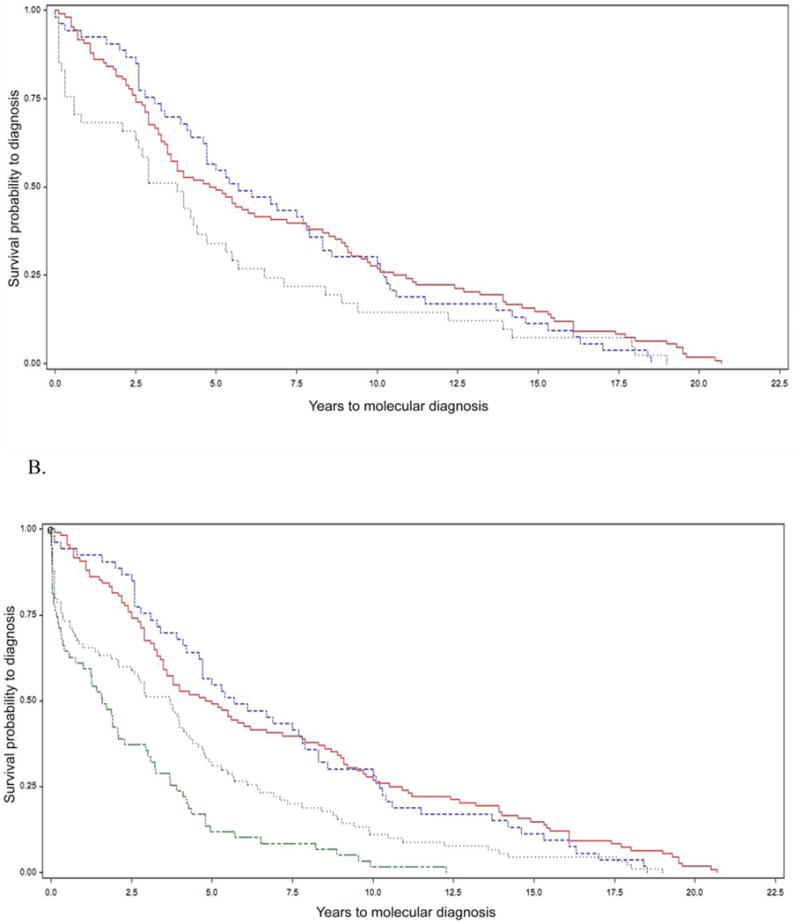
Clinical molecular testing has been available for 22q11.2 deletion syndrome (22q11.2DS) for over two decades yet under-recognition and diagnostic delays are common. To characterize the "diagnostic odyssey" in 22q11.2DS we studied 202 well-characterized unrelated adults, none ascertained through an affected relative. We used a regression model to identify clinical and demographic factors associated with length of time to molecular diagnosis. Kaplan-Meier analysis compared time to diagnosis for the molecular testing era (since 1994) and earlier birth cohorts. The results showed that the median time to molecular diagnosis of the 22q11.2 deletion was 4.7 (range 0-20.7) years. Palatal and cardiac anomalies, but not developmental delay/intellectual disability, were associated with a shorter time to molecular diagnosis. Non-European ethnicity was associated with longer time to diagnosis. Inclusion of a cohort from another 22q11.2DS center increased power to observe a significantly earlier diagnosis for patients born in the molecular testing era. Nonetheless, only a minority were diagnosed in the first year of life. On average, patients were seen in seven (range 2-15) different clinical specialty areas prior to molecular diagnosis. The findings indicate that even for those born in the molecular testing era, individuals with 22q11.2DS and their families face a diagnostic odyssey that is often prolonged, particularly in the absence of typical physical congenital features or for those of non-European ancestry. The results support educational efforts to improve clinical recognition and testing, and ultimately newborn screening as a means of maximizing early detection that would provide the best opportunity to optimize outcomes.
Keywords: DiGeorge syndrome; developmental delay; feeding difficulties; neurology; psychosis proneness; recurrent infections; scoliosis; seizures; surgical complications; velocardiofacial syndrome.
© 2018 Wiley Periodicals, Inc.
Conflict of interest statement
Figures
References
-
- Bales AM, Zaleski CA, McPherson EW. Patient and family experiences and opinions on adding 22q11 deletion syndrome to the newborn screen. Journal of Genetic Counseling. 2010;19:526–534. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
