

Germline MLH1, MSH2 and MSH6 variants in Brazilian patients with colorectal cancer and clinical features suggestive of Lynch Syndrome
- PMID: 29575718
- PMCID: PMC5943474
- DOI: 10.1002/cam4.1316
Germline MLH1, MSH2 and MSH6 variants in Brazilian patients with colorectal cancer and clinical features suggestive of Lynch Syndrome
Abstract
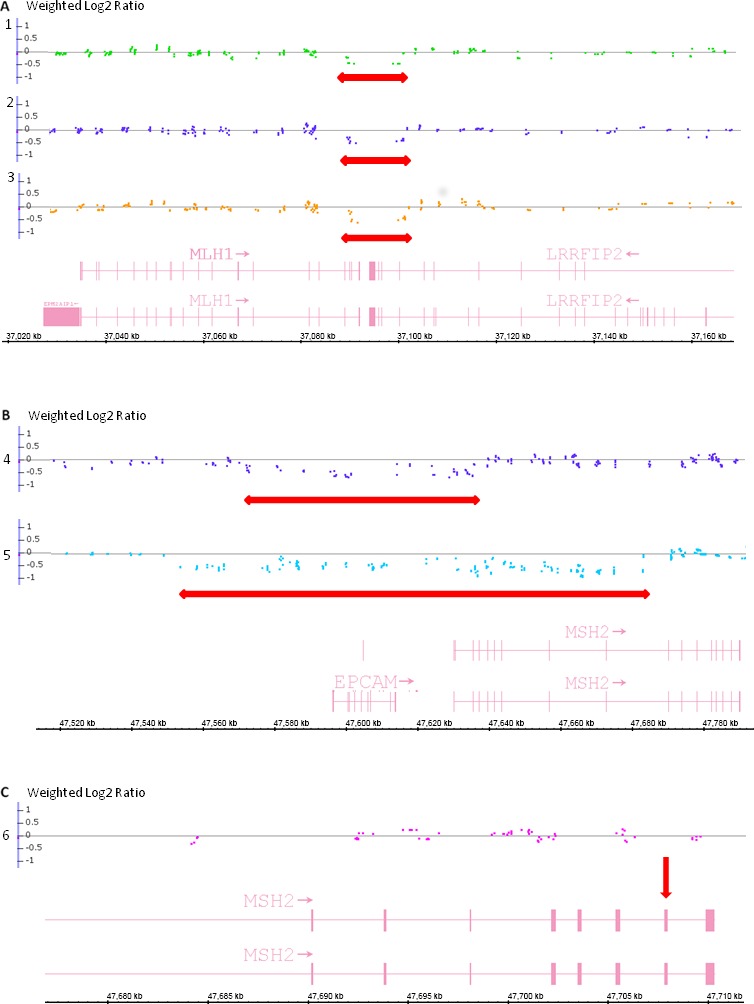
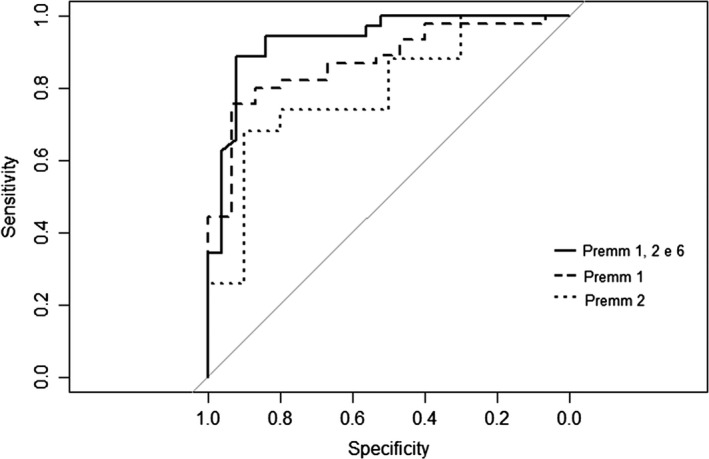
Lynch syndrome (LS) is the most common hereditary colorectal cancer syndrome, caused by germline mutations in one of the major genes involved in mismatch repair (MMR): MLH1, MSH2, MSH6 and more rarely, PMS2. Recently, germline deletions in EPCAM have been also associated to the syndrome. Most of the pathogenic MMR mutations found in LS families occur in MLH1 or MSH2. Gene variants include missense, nonsense, frameshift mutations, large genomic rearrangements and splice-site variants and most of the studies reporting the molecular characterization of LS families have been conducted outside South America. In this study, we analyzed 60 unrelated probands diagnosed with colorectal cancer and LS criteria. Testing for germline mutations and/or rearrangements in the most commonly affected MMR genes (MLH1, MSH2, EPCAM and MSH6) was done by Sanger sequencing and MLPA. Pathogenic or likely pathogenic variants were identified in MLH1 or MSH2 in 21 probands (35.0%). Of these, approximately one-third were gene rearrangements. In addition, nine variants of uncertain significance (VUS) were identified in 10 (16.6%) of the sixty probands analyzed. Other four novel variants were identified, only in MLH1. Our results suggest that MSH6 pathogenic variants are not common among Brazilian LS probands diagnosed with CRC and that MMR gene rearrangements account for a significant proportion of the germline variants in this population underscoring the need to include rearrangement analysis in the molecular testing of Brazilian individuals with suspected Lynch syndrome.
Keywords: Colorectal cancer; Lynch syndrome; MMR genes.
© 2018 The Authors. Cancer Medicine published by John Wiley & Sons Ltd.
Figures
Similar articles
-
Germline variants screening of MLH1, MSH2, MSH6 and PMS2 genes in 64 Algerian Lynch syndrome families: The first nationwide study.Ann Hum Genet. 2022 Nov;86(6):328-352. doi: 10.1111/ahg.12482. Epub 2022 Sep 8. Ann Hum Genet. 2022. PMID: 36073783
-
First description of mutational analysis of MLH1, MSH2 and MSH6 in Algerian families with suspected Lynch syndrome.Fam Cancer. 2017 Jan;16(1):57-66. doi: 10.1007/s10689-016-9917-1. Fam Cancer. 2017. PMID: 27468915
-
Universal screening for Lynch syndrome in endometrial cancers: frequency of germline mutations and identification of patients with Lynch-like syndrome.Hum Pathol. 2017 Dec;70:121-128. doi: 10.1016/j.humpath.2017.10.022. Epub 2017 Oct 28. Hum Pathol. 2017. PMID: 29107668
-
The Clinical Outcomes Among Patients Under 60 Years Old with Lynch Syndrome: Variations Based on Different Mutation Patterns.Int J Mol Sci. 2025 Apr 4;26(7):3383. doi: 10.3390/ijms26073383. Int J Mol Sci. 2025. PMID: 40244260 Free PMC article. Review.
-
Identification of Lynch Syndrome.Gastrointest Endosc Clin N Am. 2022 Jan;32(1):45-58. doi: 10.1016/j.giec.2021.09.002. Gastrointest Endosc Clin N Am. 2022. PMID: 34798986 Review.
Cited by
-
Identification of genomic variants associated with colorectal cancer heredity in indigenous populations of the Amazon.Sci Rep. 2025 Apr 26;15(1):14616. doi: 10.1038/s41598-025-87401-0. Sci Rep. 2025. PMID: 40287430 Free PMC article.
-
Association of a novel frameshift variant and a known deleterious variant in MMR genes with Lynch syndrome in Chinese families.World J Surg Oncol. 2024 Jan 27;22(1):36. doi: 10.1186/s12957-024-03309-5. World J Surg Oncol. 2024. PMID: 38280988 Free PMC article.
-
Mutation Spectrum of Cancer-Associated Genes in Patients With Early Onset of Colorectal Cancer.Front Oncol. 2019 Aug 2;9:673. doi: 10.3389/fonc.2019.00673. eCollection 2019. Front Oncol. 2019. PMID: 31428572 Free PMC article.
-
Correlation of mismatch repair deficiency with clinicopathological features and programmed death-ligand 1 expression in thyroid carcinoma.Endocrine. 2022 Jun;76(3):660-670. doi: 10.1007/s12020-022-03031-w. Epub 2022 Apr 2. Endocrine. 2022. PMID: 35366156
-
Prevalence and Spectrum of Predisposition Genes With Germline Mutations Among Chinese Patients With Bowel Cancer.Front Genet. 2022 Jan 27;12:755629. doi: 10.3389/fgene.2021.755629. eCollection 2021. Front Genet. 2022. PMID: 35154239 Free PMC article.
References
-
- Lynch, H. T. , and Smyrk T.. 1996. Hereditary nonpolyposis colorectal cancer (Lynch Syndrome). An updated review. Cancer 78:1149–1167. - PubMed
-
- Lynch, H. , and de la Chapelle A.. 2003. Hereditary colorectal cancer. N. Engl. J. Med. 348:919–932. - PubMed
-
- Kovacs, M. E. , Papp J., Szentirmay Z., Otto S., and Olah E.. 2009. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum. Mutat. 30:197–203. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
