

Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns
- PMID: 29576454
- PMCID: PMC6643283
- DOI: 10.1016/j.cell.2018.02.037
Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns
Abstract
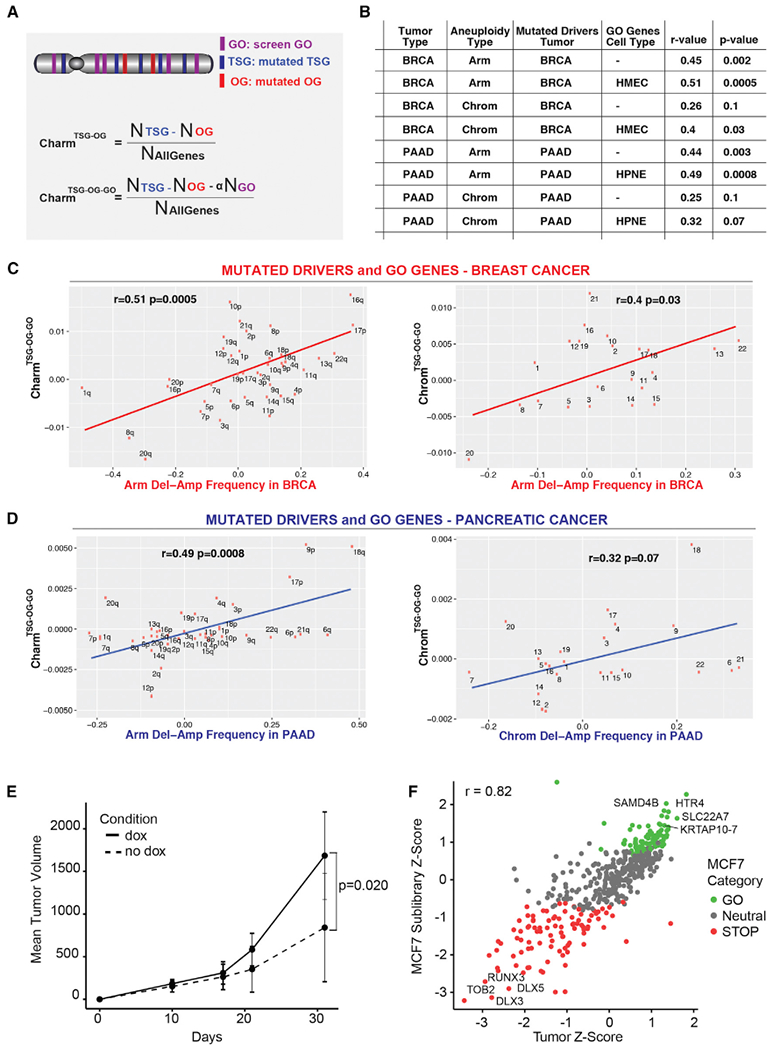
Genomics has provided a detailed structural description of the cancer genome. Identifying oncogenic drivers that work primarily through dosage changes is a current challenge. Unrestrained proliferation is a critical hallmark of cancer. We constructed modular, barcoded libraries of human open reading frames (ORFs) and performed screens for proliferation regulators in multiple cell types. Approximately 10% of genes regulate proliferation, with most performing in an unexpectedly highly tissue-specific manner. Proliferation drivers in a given cell type showed specific enrichment in somatic copy number changes (SCNAs) from cognate tumors and helped predict aneuploidy patterns in those tumors, implying that tissue-type-specific genetic network architectures underlie SCNA and driver selection in different cancers. In vivo screening confirmed these results. We report a substantial contribution to the catalog of SCNA-associated cancer drivers, identifying 147 amplified and 107 deleted genes as potential drivers, and derive insights about the genetic network architecture of aneuploidy in tumors.
Keywords: KRTAP; ORF screens; SCNA; aneuploidy; cancer drivers; gain of function screens; genetic screens; proliferation; tissue specificity.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
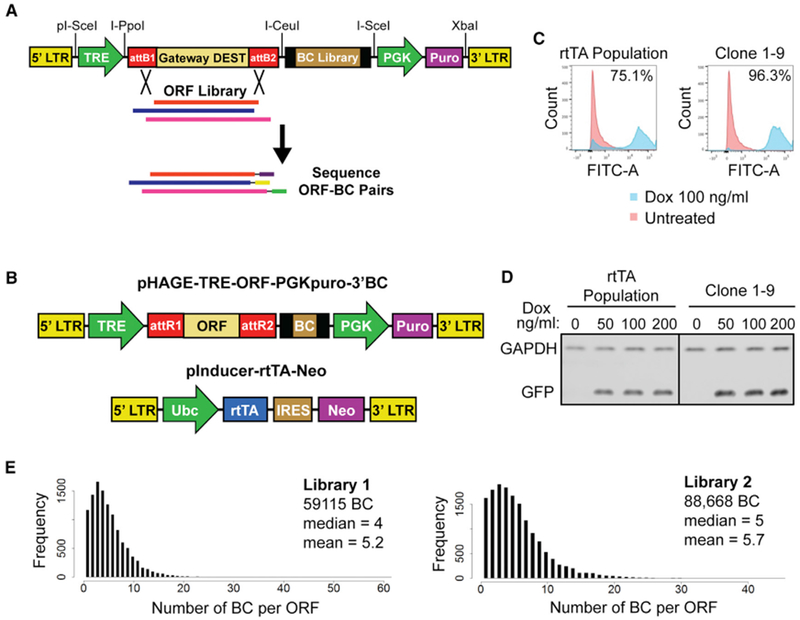
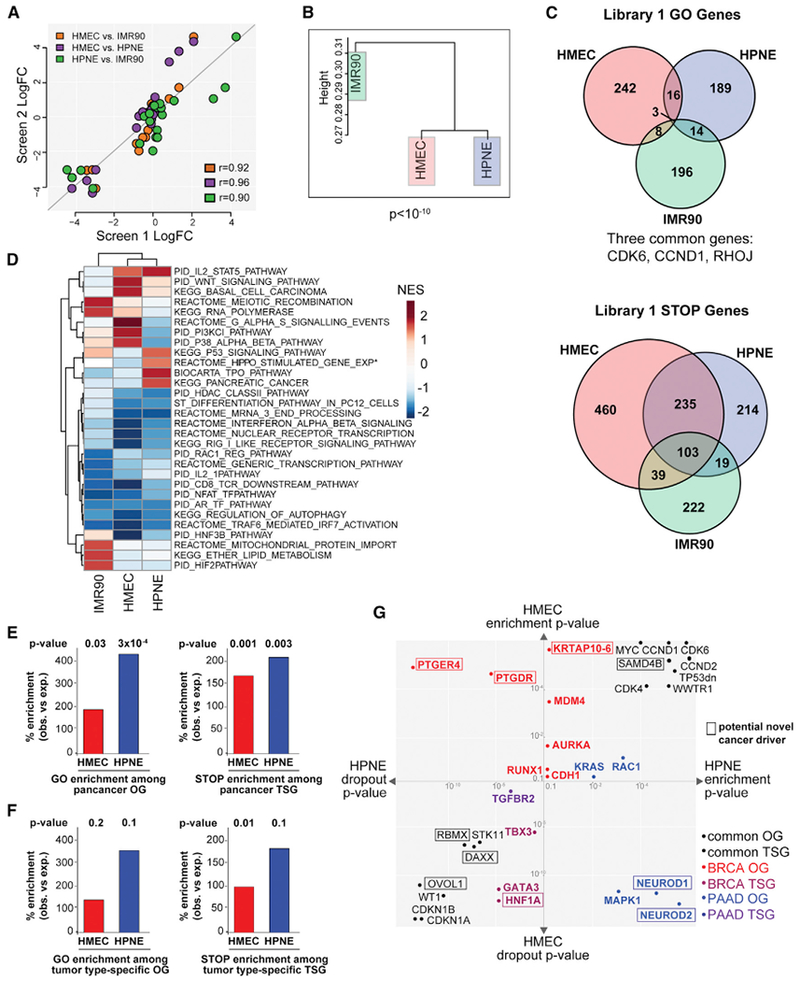
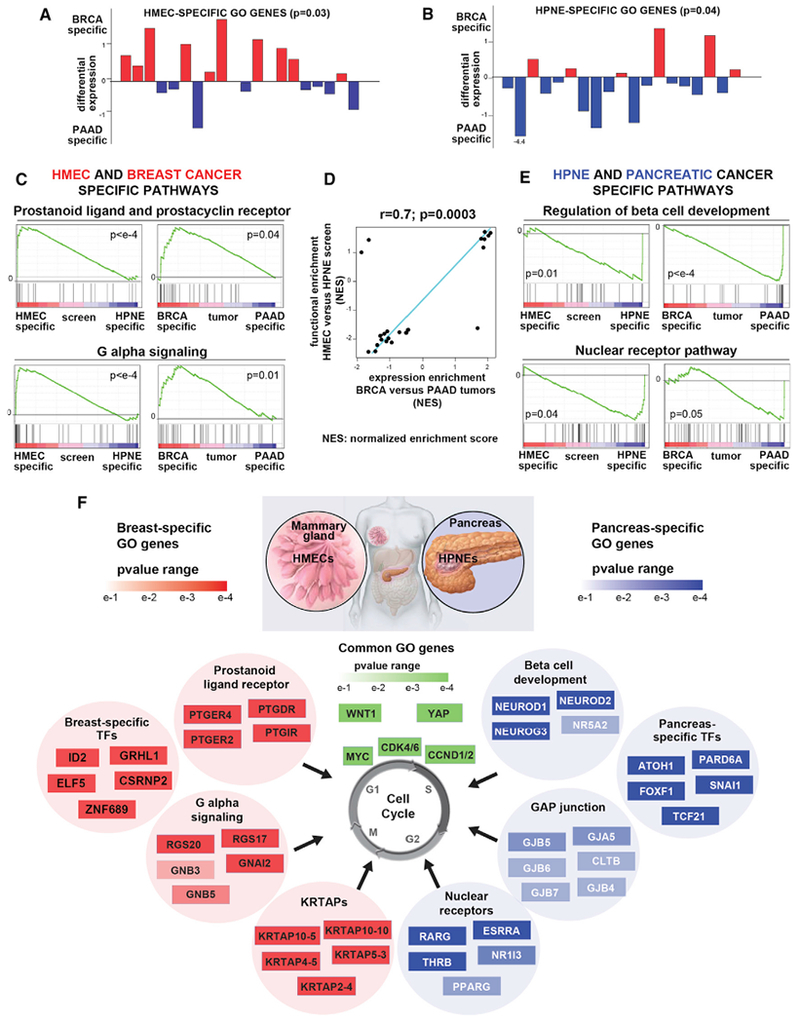

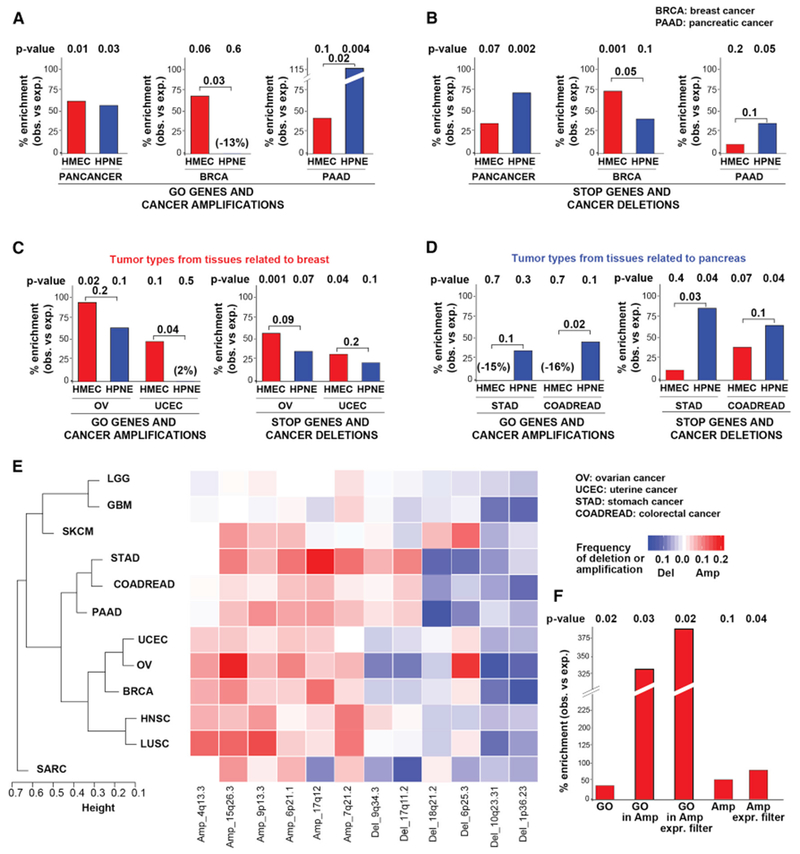

Comment in
-
Tissue-specific oncogene screens.Nat Methods. 2018 Jun;15(6):408. doi: 10.1038/s41592-018-0027-x. Nat Methods. 2018. PMID: 29855584 No abstract available.
References
-
- Asselin-Labat M-L, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC, Hartley L, Robb L, Grosveld FG, van der Wees J, et al. (2007). Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol 9, 201–209. - PubMed
-
- Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, Miller DK, Christ AN, Bruxner TJC, Quinn MC, et al.; Australian Pancreatic Cancer Genome Initiative (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
