

A mechanistic pan-cancer pathway model informed by multi-omics data interprets stochastic cell fate responses to drugs and mitogens
- PMID: 29579036
- PMCID: PMC5886578
- DOI: 10.1371/journal.pcbi.1005985
A mechanistic pan-cancer pathway model informed by multi-omics data interprets stochastic cell fate responses to drugs and mitogens
Erratum in
-
Correction: A mechanistic pan-cancer pathway model informed by multi-omics data interprets stochastic cell fate responses to drugs and mitogens.PLoS Comput Biol. 2018 Jul 9;14(7):e1006189. doi: 10.1371/journal.pcbi.1006189. eCollection 2018 Jul. PLoS Comput Biol. 2018. PMID: 29985911 Free PMC article.
Abstract
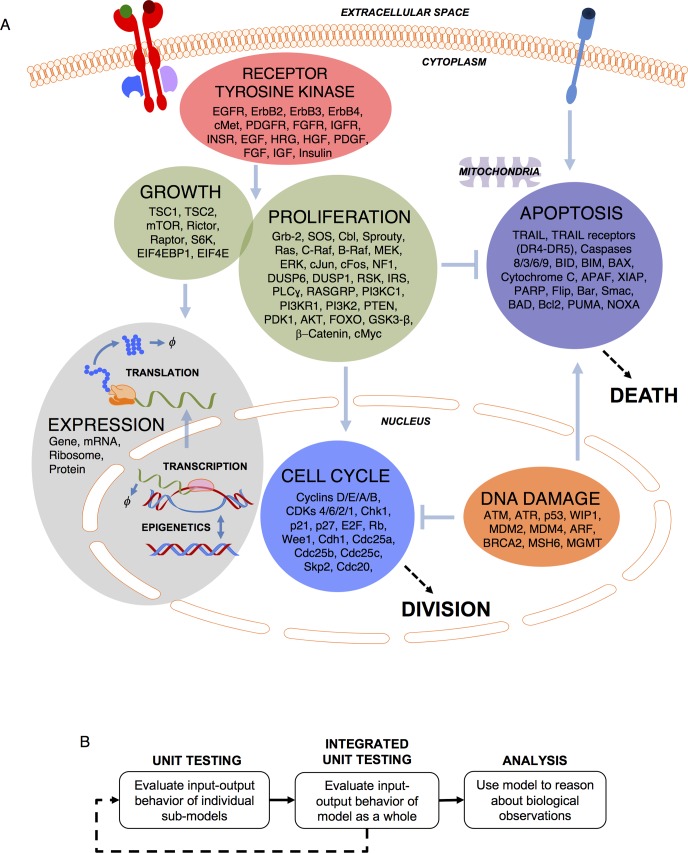
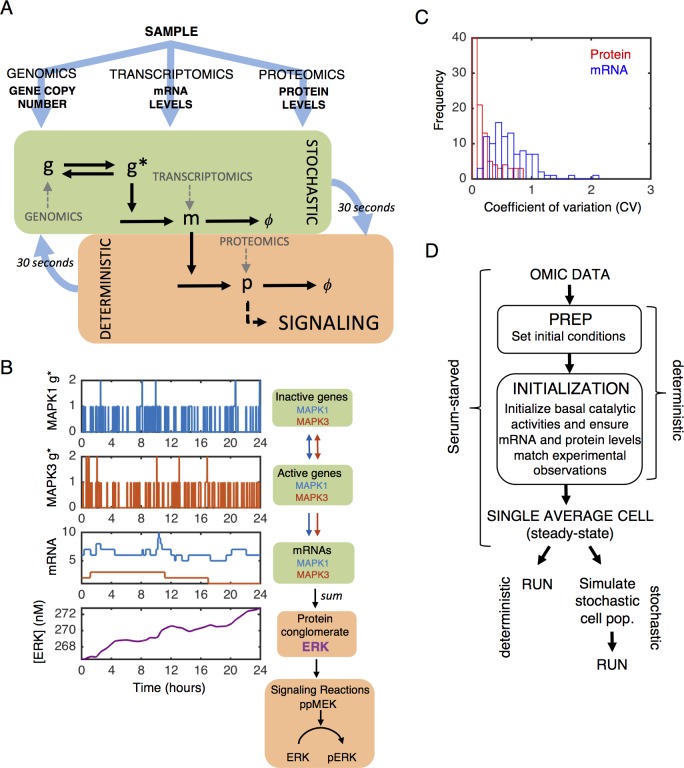
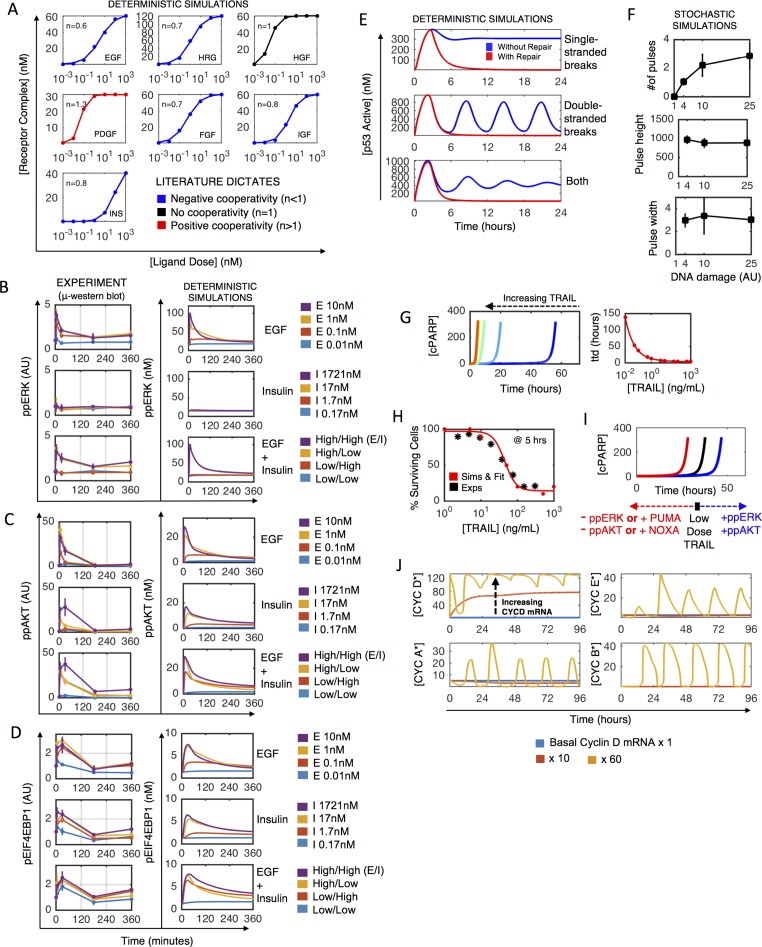
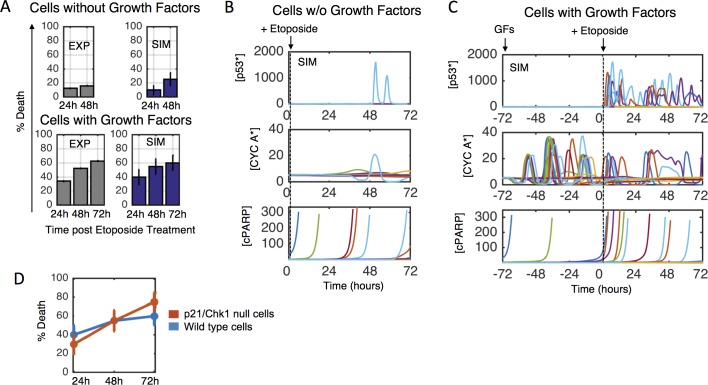
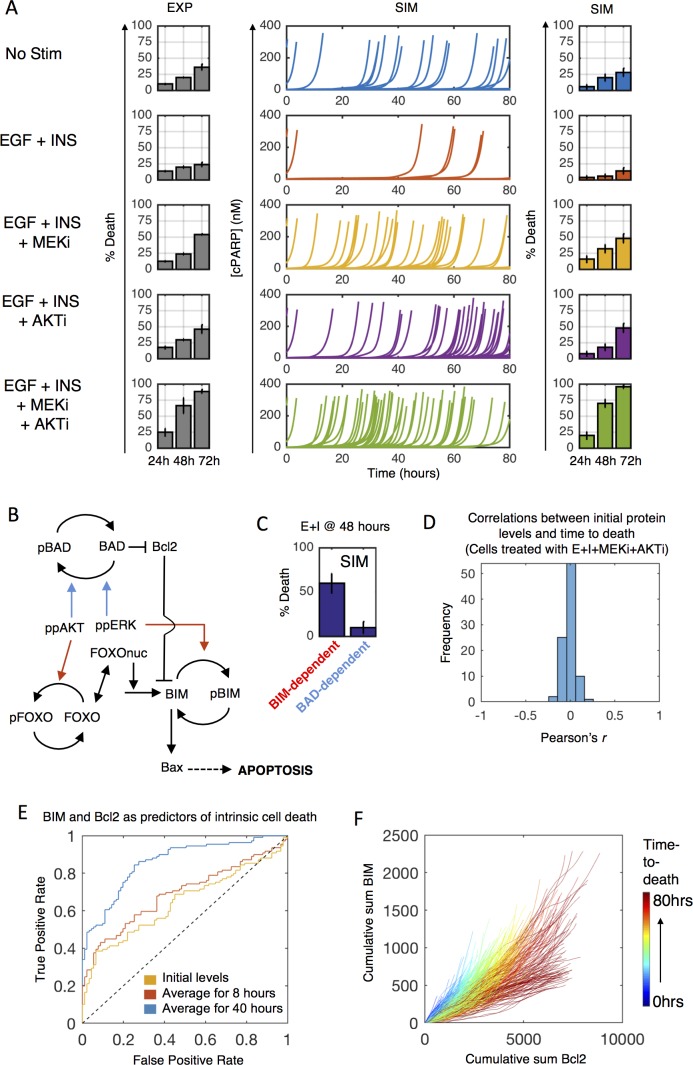
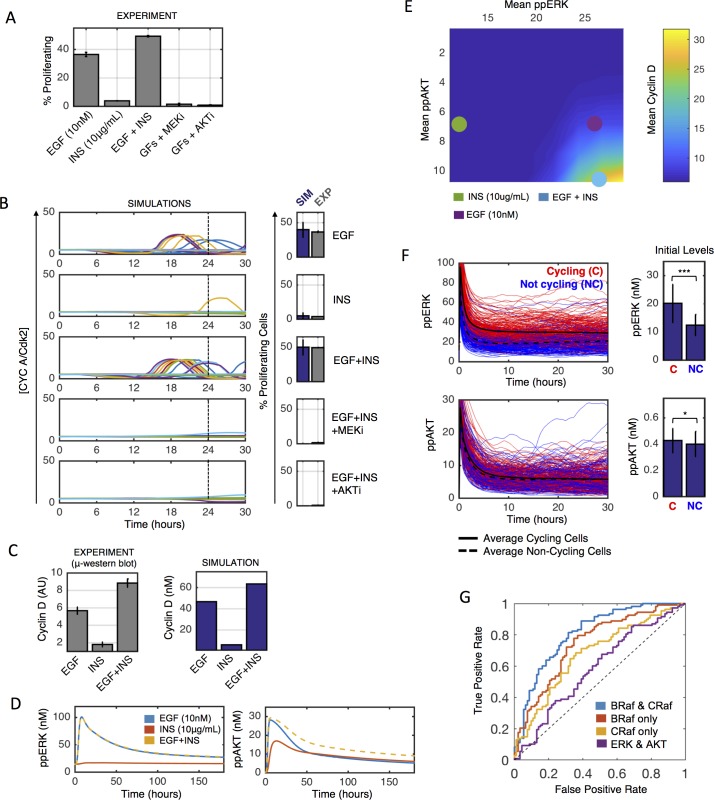
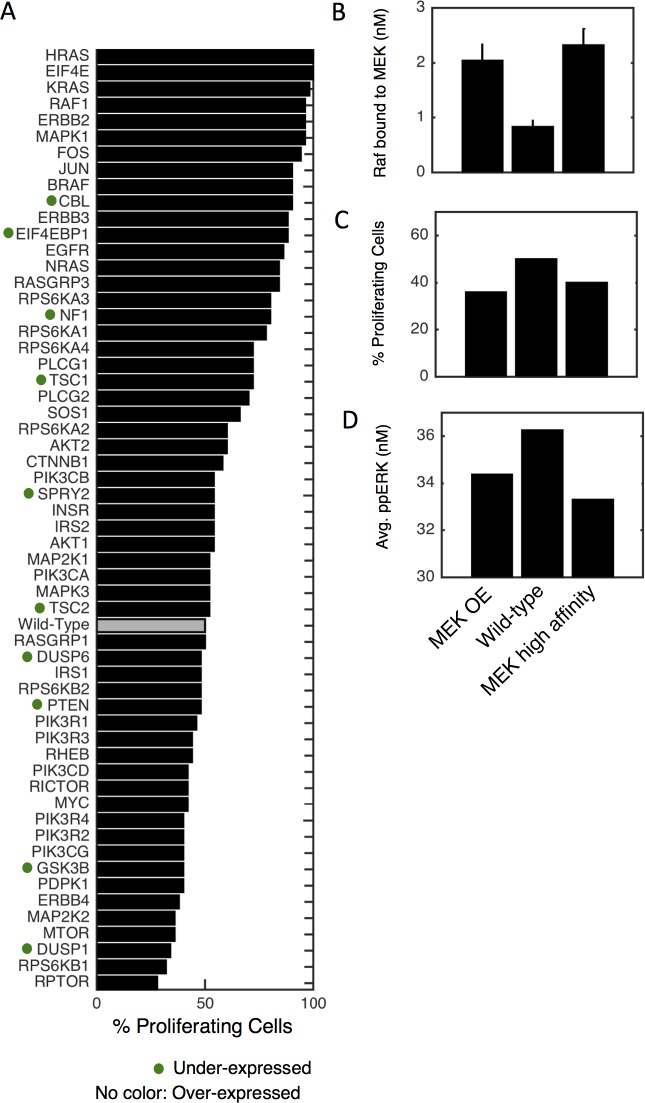
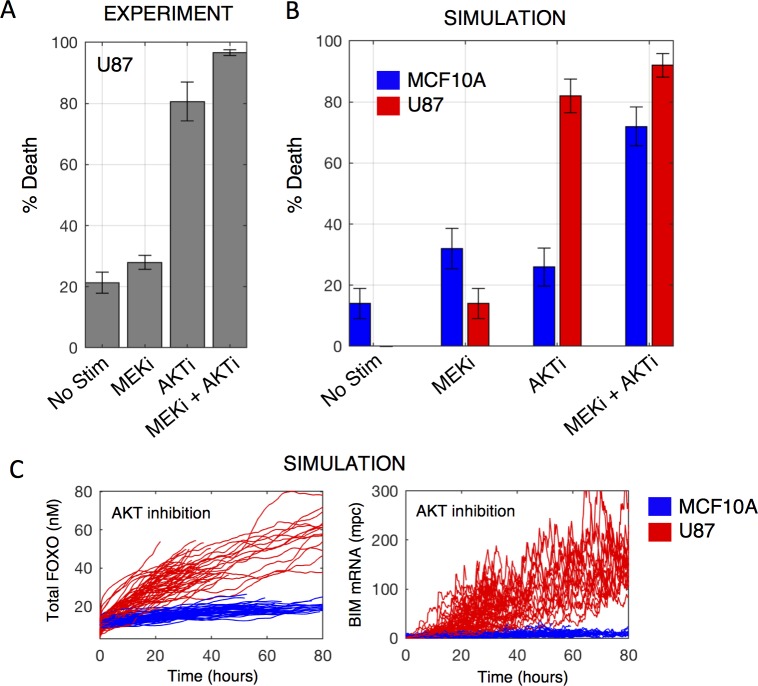
Most cancer cells harbor multiple drivers whose epistasis and interactions with expression context clouds drug and drug combination sensitivity prediction. We constructed a mechanistic computational model that is context-tailored by omics data to capture regulation of stochastic proliferation and death by pan-cancer driver pathways. Simulations and experiments explore how the coordinated dynamics of RAF/MEK/ERK and PI-3K/AKT kinase activities in response to synergistic mitogen or drug combinations control cell fate in a specific cellular context. In this MCF10A cell context, simulations suggest that synergistic ERK and AKT inhibitor-induced death is likely mediated by BIM rather than BAD, which is supported by prior experimental studies. AKT dynamics explain S-phase entry synergy between EGF and insulin, but simulations suggest that stochastic ERK, and not AKT, dynamics seem to drive cell-to-cell proliferation variability, which in simulations is predictable from pre-stimulus fluctuations in C-Raf/B-Raf levels. Simulations suggest MEK alteration negligibly influences transformation, consistent with clinical data. Tailoring the model to an alternate cell expression and mutation context, a glioma cell line, allows prediction of increased sensitivity of cell death to AKT inhibition. Our model mechanistically interprets context-specific landscapes between driver pathways and cell fates, providing a framework for designing more rational cancer combination therapy.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344: 1031–7. doi: 10.1056/NEJM200104053441401 - DOI - PubMed
-
- Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17: 2639–48. doi: 10.1200/JCO.1999.17.9.2639 - DOI - PubMed
-
- Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20: 719–26. doi: 10.1200/JCO.2002.20.3.719 - DOI - PubMed
-
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464: 427–30. doi: 10.1038/nature08902 - DOI - PMC - PubMed
-
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367: 107–14. doi: 10.1056/NEJMoa1203421 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
