

Review
doi: 10.1093/bfgp/ely009.
Quantitative single-cell transcriptomics
Affiliations
- PMID: 29579145
- PMCID: PMC6063296
- DOI: 10.1093/bfgp/ely009
Item in Clipboard
Review
Quantitative single-cell transcriptomics
Brief Funct Genomics.
.
Abstract
Single-cell RNA sequencing (scRNA-seq) is currently transforming our understanding of biology, as it is a powerful tool to resolve cellular heterogeneity and molecular networks. Over 50 protocols have been developed in recent years and also data processing and analyzes tools are evolving fast. Here, we review the basic principles underlying the different experimental protocols and how to benchmark them. We also review and compare the essential methods to process scRNA-seq data from mapping, filtering, normalization and batch corrections to basic differential expression analysis. We hope that this helps to choose appropriate experimental and computational methods for the research question at hand.
Figures
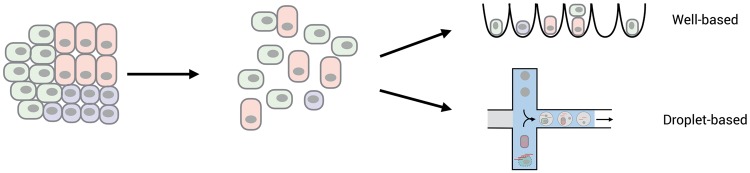
Single-cell isolation. Almost all scRNA-seq methods require to dissociate cells to make a single-cell suspension. To what extend this suspension represents the cellular composition and the expression patterns of the original population is a major challenge for many tissues. In addition, using frozen samples as starting material is often not possible and can be overcome by making a suspension of nuclei instead of cells (not shown). A major difference among scRNA-seq methods is whether single wells are distributed in a controlled fashion among wells, e.g. by FACS, or randomly distributed across containers e.g using microdroplets.
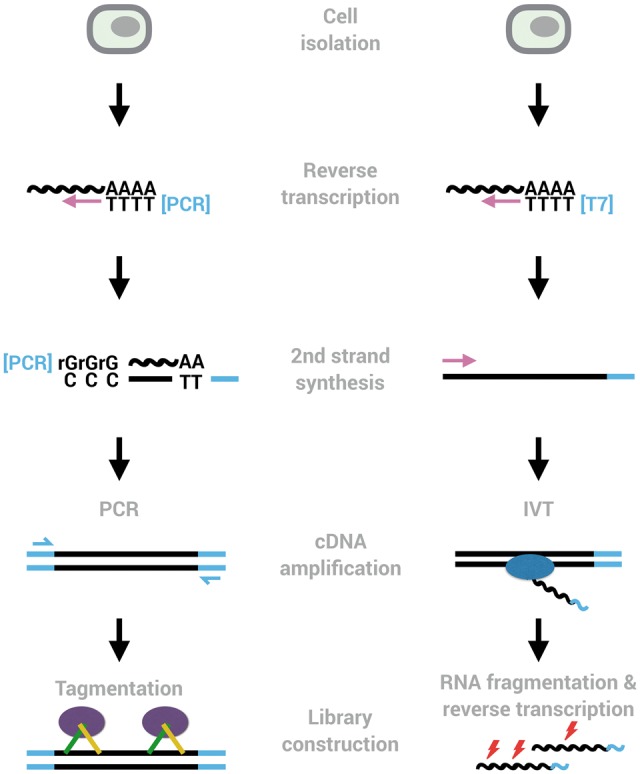
Two common workflows of generating scRNA-seq libraries. Many methods use oligo-dT priming, template switching, pre-amplification by PCR and tagmentation to generate libraries (left). The major other amplification method amplifies cDNA linearly using in vitro transcription (right). Early barcodes and UMIs can be introduced into the primers used for reverse transcription or for second-strand synthesis, allowing to pool reactions from many cells and to identify amplified molecules, respectively.
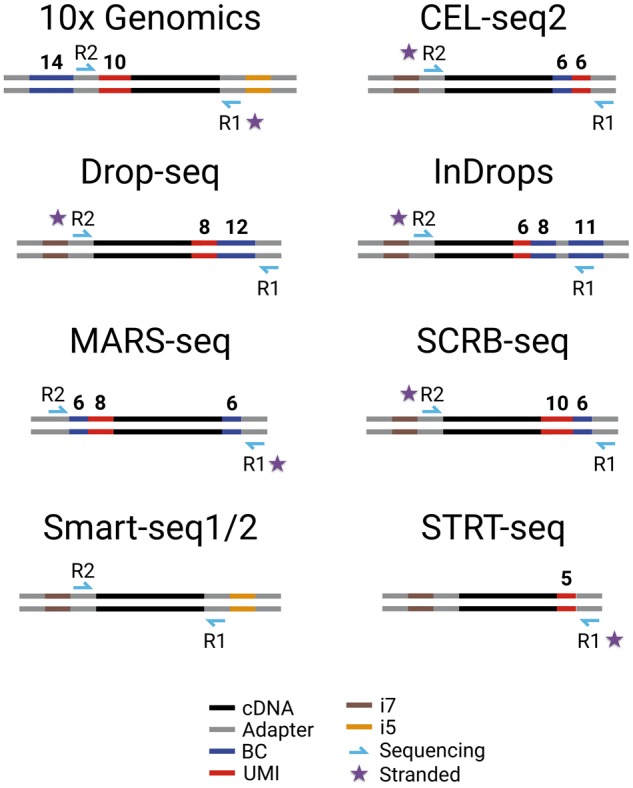
Overview of commonly used scRNA-seq libraries. Shown are the length and position of barcodes that distinguish cells [Barcode (BC)], UMIs, sequencing primers, Illumina indices (i5, i7) and adapter sequences needed for PCR, tagmentation and sequencing. Note that except for Smart-seq1/2, all methods contain BCs and UMIs and preserve the strand information (star). As a consequence, only Smart-seq1/2 among the shown libraries provides full-length information.
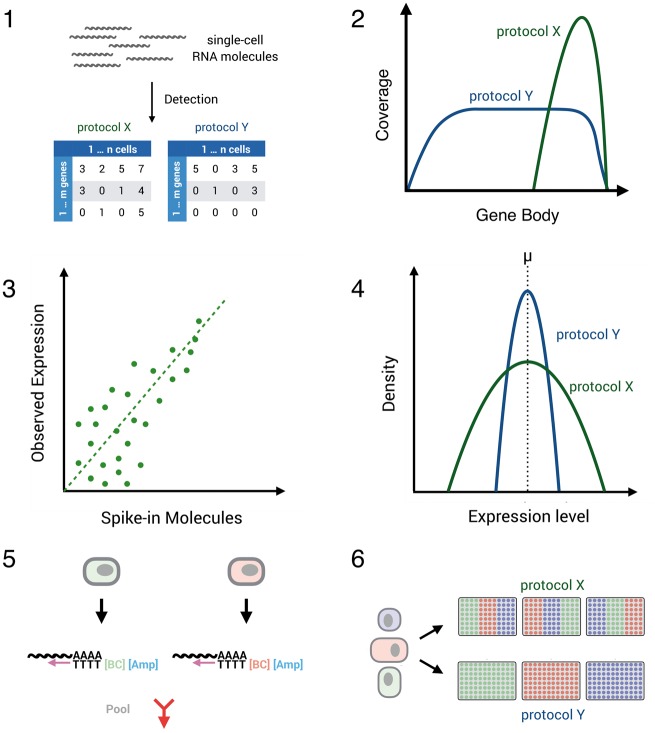
Comparing scRNA-seq protocol performance. Several aspects determine the technical performance of a scRNA-seq protocol: (1) sensitivity of protocols to detect mRNA transcripts can be defined by the number of genes/transcripts (UMIs) per cell detected above stochastic noise. (2) Coverage of transcripts: with Smart-seq1/2, ideally the full length of the transcript is covered. Conversely, early barcoding and UMI-methods enrich for the 3′ and 5′ prime end of the sequences. (3) Accuracy of estimated expression levels reflect the correlation of known transcript concentrations and measured transcript expression. Notably, this correlation also depends on the sensitivity and precision of a method. (4) Precision of estimated expression levels reflects the measurement error of expression in single cells and depends on sensitivity and amplification noise. The latter is essentially abolished by UMIs. (5) The throughput of a method depends on the cell isolation method and on the costs per cell, which are strongly reduced by the depicted early barcoding (6) Batch effects of library generation can be a decisive factor for interpreting results, and methods that allow a balanced experimental design have a decisive advantage in this respect.
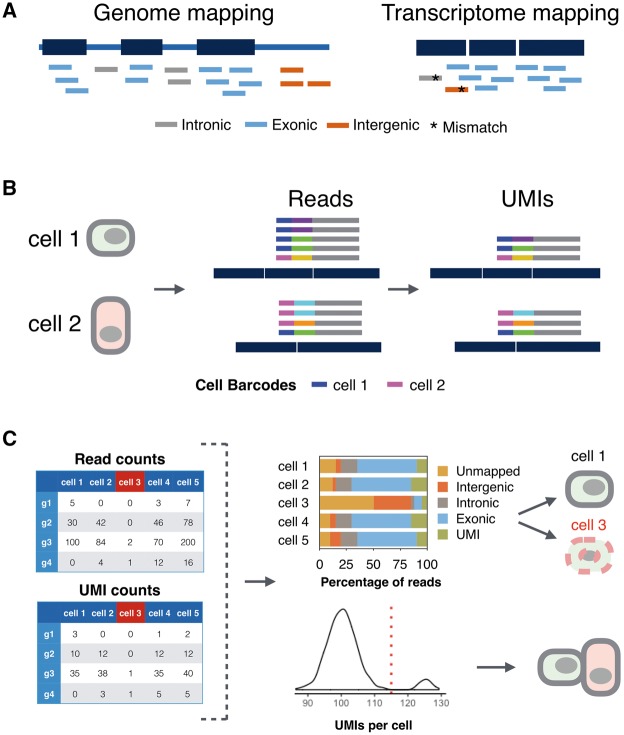
Processing scRNA-seq data. (A) The mapping strategy is a crucial step in scRNA-seq data processing that influences the end results. Reads are mapped to reference genome (left) or transcriptome (right). Short reads generated from introns, exons and intergenic regions are colored gray, blue and orange, respectively. As intronic and intergenic reads can be mapped wrongly (asterisk), mapping only to the transcriptome is not recommended. (B) Correctly assigning cDNA reads (gray) with early barcodes (blue and magenta) and UMIs (other colors) to two genes requires to control for sequencing errors in barcodes and UMIs. (C) Filtering ‘bad’ cells and doublets based on mapping reads and counting UMIs, respectively. Bad cells, such as Cell 3 in this example, have a low percentage of exonic reads (upper panel) and a low correlation to other cells (not shown). Doublets have on average a transcript count that is twice the population average (lower panel).
References
-
- Reinius B, Sandberg R.. Random monoallelic expression of autosomal genes: stochastic transcription and allele-level regulation. Nat Rev Genet 2015;16:653–64. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
