

A Bayesian framework for multiple trait colocalization from summary association statistics
- PMID: 29579179
- PMCID: PMC6061859
- DOI: 10.1093/bioinformatics/bty147
A Bayesian framework for multiple trait colocalization from summary association statistics
Abstract
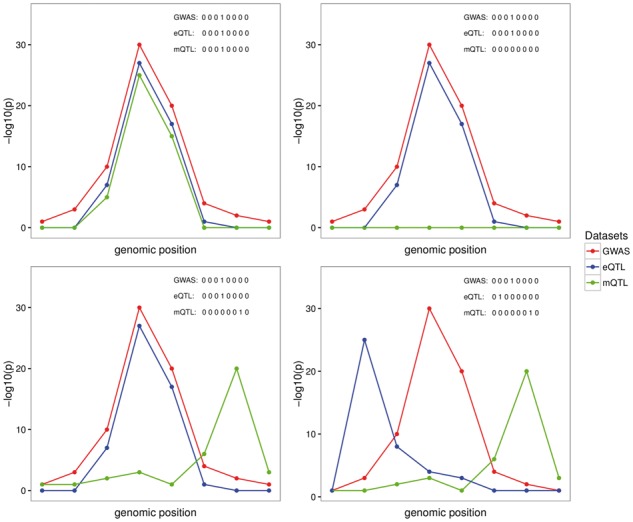
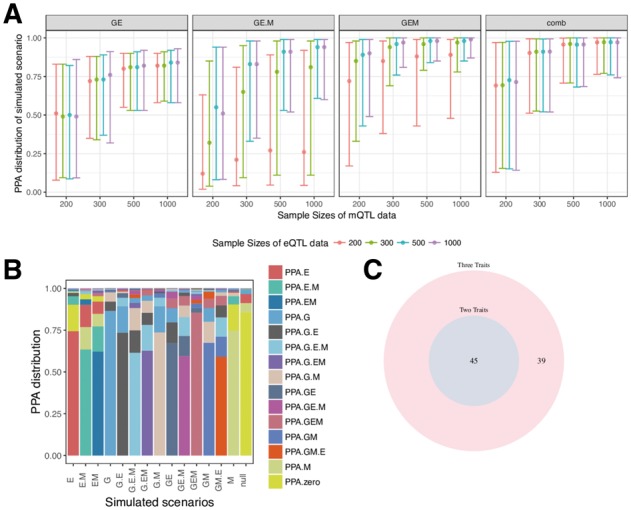
Motivation: Most genetic variants implicated in complex diseases by genome-wide association studies (GWAS) are non-coding, making it challenging to understand the causative genes involved in disease. Integrating external information such as quantitative trait locus (QTL) mapping of molecular traits (e.g. expression, methylation) is a powerful approach to identify the subset of GWAS signals explained by regulatory effects. In particular, expression QTLs (eQTLs) help pinpoint the responsible gene among the GWAS regions that harbor many genes, while methylation QTLs (mQTLs) help identify the epigenetic mechanisms that impact gene expression which in turn affect disease risk. In this work, we propose multiple-trait-coloc (moloc), a Bayesian statistical framework that integrates GWAS summary data with multiple molecular QTL data to identify regulatory effects at GWAS risk loci.
Results: We applied moloc to schizophrenia (SCZ) and eQTL/mQTL data derived from human brain tissue and identified 52 candidate genes that influence SCZ through methylation. Our method can be applied to any GWAS and relevant functional data to help prioritize disease associated genes. Availability and implementation: moloc is available for download as an R package (https://github.com/clagiamba/moloc). We also developed a web site to visualize the biological findings (icahn.mssm.edu/moloc). The browser allows searches by gene, methylation probe and scenario of interest.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
References
Publication types
MeSH terms
Grants and funding
- P50 MH096891/MH/NIMH NIH HHS/United States
- U01 CA194393/CA/NCI NIH HHS/United States
- R01 MH075916/MH/NIMH NIH HHS/United States
- R01 MH109677/MH/NIMH NIH HHS/United States
- P50 MH084053/MH/NIMH NIH HHS/United States
- R01 HG009120/HG/NHGRI NIH HHS/United States
- HHSN271201300031C/MH/NIMH NIH HHS/United States
- P50 MH066392/MH/NIMH NIH HHS/United States
- R01 MH080405/MH/NIMH NIH HHS/United States
- R01 MH085542/MH/NIMH NIH HHS/United States
- R01 AG050986/AG/NIA NIH HHS/United States
- R37 MH057881/MH/NIMH NIH HHS/United States
- T32 NS048004/NS/NINDS NIH HHS/United States
- R01 HG006399/HG/NHGRI NIH HHS/United States
- R01 MH106842/MH/NIMH NIH HHS/United States
- I01 BX002395/BX/BLRD VA/United States
- R01 MH097276/MH/NIMH NIH HHS/United States
- P01 AG002219/AG/NIA NIH HHS/United States
- R01 MH093725/MH/NIMH NIH HHS/United States
- P50 AG005138/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
