

Truncating SLC5A7 mutations underlie a spectrum of dominant hereditary motor neuropathies
- PMID: 29582019
- PMCID: PMC5866402
- DOI: 10.1212/NXG.0000000000000222
Truncating SLC5A7 mutations underlie a spectrum of dominant hereditary motor neuropathies
Abstract
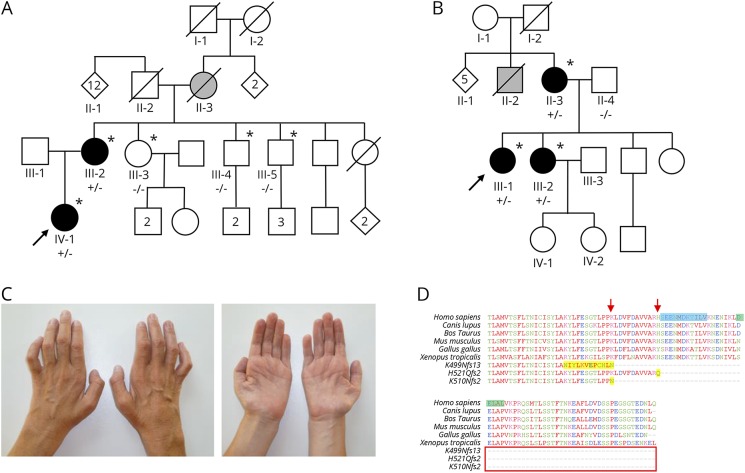
Objective: To identify the genetic cause of disease in 2 previously unreported families with forms of distal hereditary motor neuropathies (dHMNs).
Methods: The first family comprises individuals affected by dHMN type V, which lacks the cardinal clinical feature of vocal cord paralysis characteristic of dHMN-VII observed in the second family. Next-generation sequencing was performed on the proband of each family. Variants were annotated and filtered, initially focusing on genes associated with neuropathy. Candidate variants were further investigated and confirmed by dideoxy sequence analysis and cosegregation studies. Thorough patient phenotyping was completed, comprising clinical history, examination, and neurologic investigation.
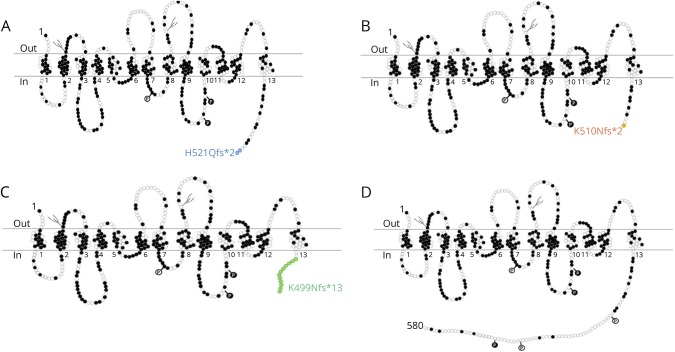
Results: dHMNs are a heterogeneous group of peripheral motor neuron disorders characterized by length-dependent neuropathy and progressive distal limb muscle weakness and wasting. We previously reported a dominant-negative frameshift mutation located in the concluding exon of the SLC5A7 gene encoding the choline transporter (CHT), leading to protein truncation, as the likely cause of dominantly-inherited dHMN-VII in an extended UK family. In this study, our genetic studies identified distinct heterozygous frameshift mutations located in the last coding exon of SLC5A7, predicted to result in the truncation of the CHT C-terminus, as the likely cause of the condition in each family.
Conclusions: This study corroborates C-terminal CHT truncation as a cause of autosomal dominant dHMN, confirming upper limb predominating over lower limb involvement, and broadening the clinical spectrum arising from CHT malfunction.
Figures
References
-
- Apparsundaram S, Ferguson SM, George AL Jr, Blakely RD. Molecular cloning of a human, hemicholinium-3-sensitive choline transporter. Biochem Biophys Res Commun 2000;276:862–867. - PubMed
-
- Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry 2012;83:6–14. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources