

Population genomics of Fusarium graminearum reveals signatures of divergent evolution within a major cereal pathogen
- PMID: 29584736
- PMCID: PMC5870968
- DOI: 10.1371/journal.pone.0194616
Population genomics of Fusarium graminearum reveals signatures of divergent evolution within a major cereal pathogen
Abstract
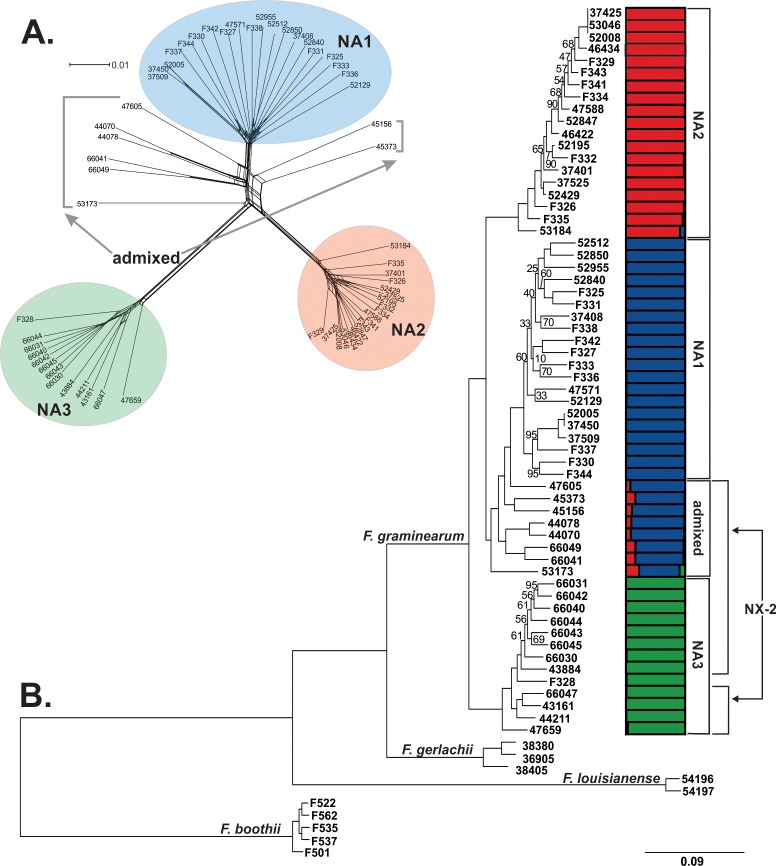
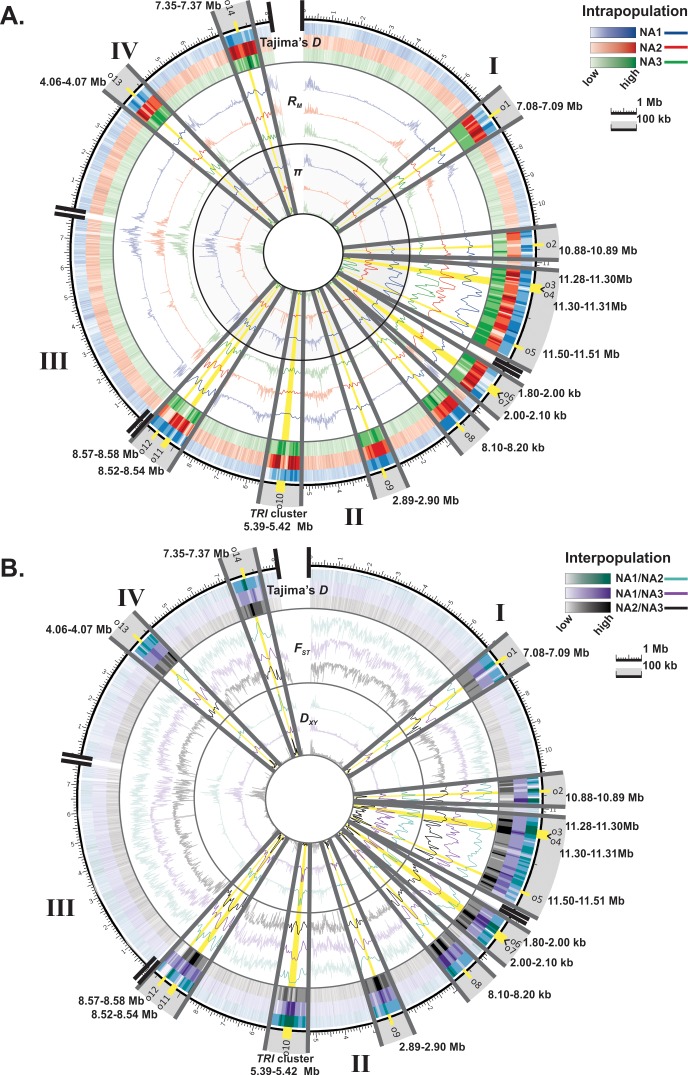
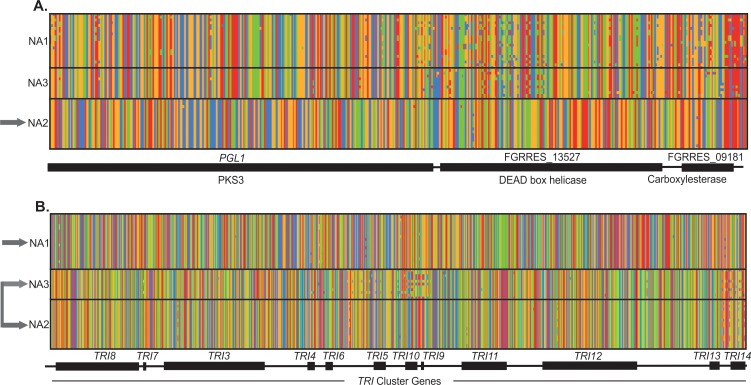
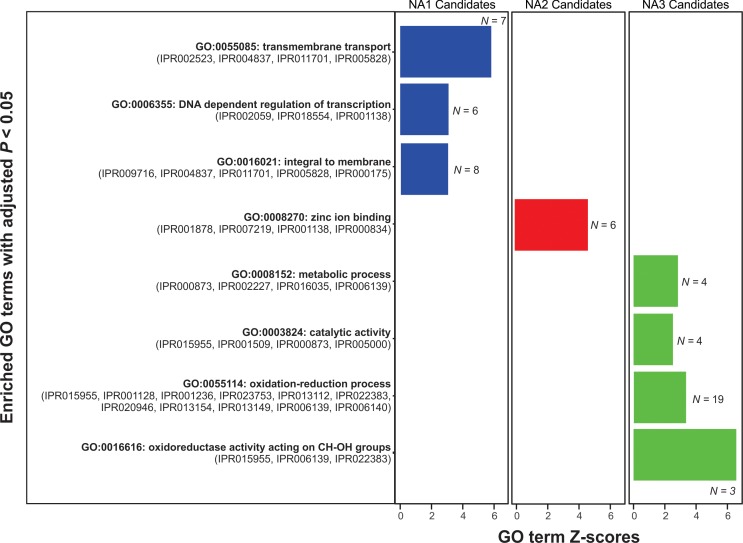
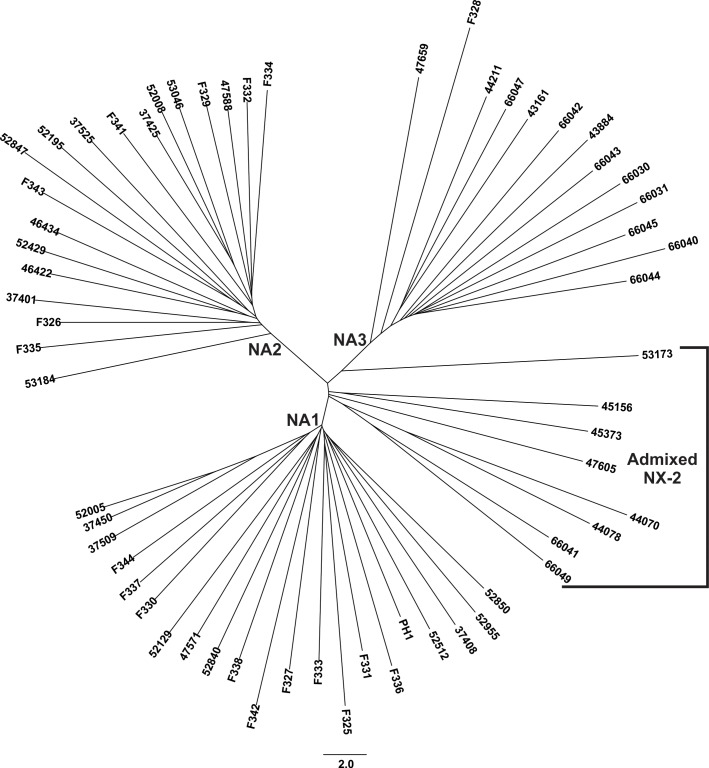
The cereal pathogen Fusarium graminearum is the primary cause of Fusarium head blight (FHB) and a significant threat to food safety and crop production. To elucidate population structure and identify genomic targets of selection within major FHB pathogen populations in North America we sequenced the genomes of 60 diverse F. graminearum isolates. We also assembled the first pan-genome for F. graminearum to clarify population-level differences in gene content potentially contributing to pathogen diversity. Bayesian and phylogenomic analyses revealed genetic structure associated with isolates that produce the novel NX-2 mycotoxin, suggesting a North American population that has remained genetically distinct from other endemic and introduced cereal-infecting populations. Genome scans uncovered distinct signatures of selection within populations, focused in high diversity, frequently recombining regions. These patterns suggested selection for genomic divergence at the trichothecene toxin gene cluster and thirteen additional regions containing genes potentially involved in pathogen specialization. Gene content differences further distinguished populations, in that 121 genes showed population-specific patterns of conservation. Genes that differentiated populations had predicted functions related to pathogenesis, secondary metabolism and antagonistic interactions, though a subset had unique roles in temperature and light sensitivity. Our results indicated that F. graminearum populations are distinguished by dozens of genes with signatures of selection and an array of dispensable accessory genes, suggesting that FHB pathogen populations may be equipped with different traits to exploit the agroecosystem. These findings provide insights into the evolutionary processes and genomic features contributing to population divergence in plant pathogens, and highlight candidate genes for future functional studies of pathogen specialization across evolutionarily and ecologically diverse fungi.
Conflict of interest statement
Figures

References
-
- Goswami RS, Kistler HC (2004) Heading for disaster: Fusarium graminearum on cereal crops. Molecular Plant Pathology 5: 515–525. doi: 10.1111/j.1364-3703.2004.00252.x - DOI - PubMed
-
- McMullen M, Bergstrom G, De Wolf E, Dill-Macky R, Hershman D, et al. (2012) A unified effort to fight an enemy of wheat and barley: Fusarium head blight. Plant Disease 96: 1712–1728. - PubMed
-
- Desjardins A, Proctor R (2007) Molecular biology of Fusarium mycotoxins. International Journal of Food Microbiology 119: 47–50. doi: 10.1016/j.ijfoodmicro.2007.07.024 - DOI - PubMed
-
- Dubin H, Gilchrist L, Reeves L, McNab A (2002) Fusarium head blight: global status and prospects In: Curtis BR, S; Macpherson HG;, editor. Bread wheat: improvement and production. Rome, Italy: Food and Agriculture Organization of the United Nations.
-
- Nganje WE, Kaitibie S, Wilson WW, Leistritz FL, Bangsund DA (2004) Economic impacts of Fusarium head blight in wheat and barley: 1993–2001. Agribusiness and Applied Economics Report No 538. ND, US: Department of Agribusiness and Applied Economics, North Dakota State University.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
