

Learned protein embeddings for machine learning
- PMID: 29584811
- PMCID: PMC6061698
- DOI: 10.1093/bioinformatics/bty178
Learned protein embeddings for machine learning
Erratum in
-
Learned protein embeddings for machine learning.Bioinformatics. 2018 Dec 1;34(23):4138. doi: 10.1093/bioinformatics/bty455. Bioinformatics. 2018. PMID: 29933431 Free PMC article. No abstract available.
Abstract
Motivation: Machine-learning models trained on protein sequences and their measured functions can infer biological properties of unseen sequences without requiring an understanding of the underlying physical or biological mechanisms. Such models enable the prediction and discovery of sequences with optimal properties. Machine-learning models generally require that their inputs be vectors, and the conversion from a protein sequence to a vector representation affects the model's ability to learn. We propose to learn embedded representations of protein sequences that take advantage of the vast quantity of unmeasured protein sequence data available. These embeddings are low-dimensional and can greatly simplify downstream modeling.
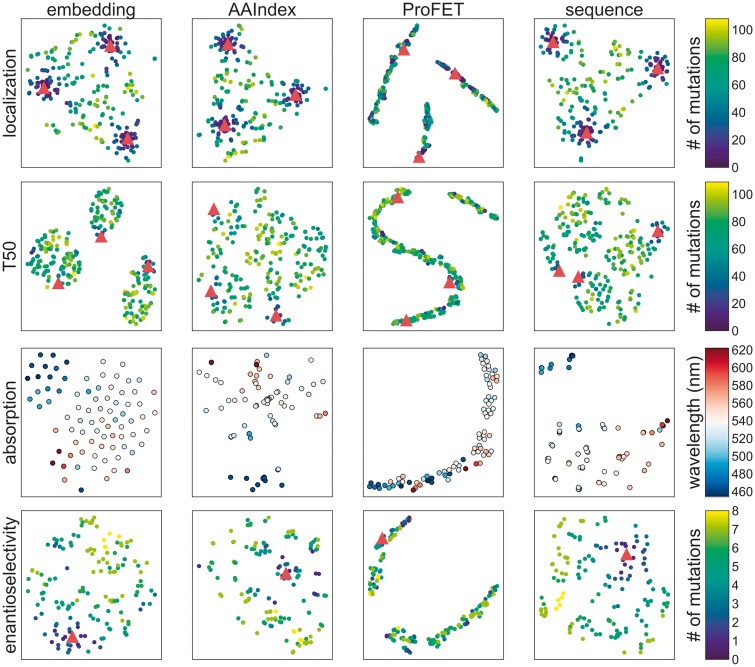
Results: The predictive power of Gaussian process models trained using embeddings is comparable to those trained on existing representations, which suggests that embeddings enable accurate predictions despite having orders of magnitude fewer dimensions. Moreover, embeddings are simpler to obtain because they do not require alignments, structural data, or selection of informative amino-acid properties. Visualizing the embedding vectors shows meaningful relationships between the embedded proteins are captured.
Availability and implementation: The embedding vectors and code to reproduce the results are available at https://github.com/fhalab/embeddings_reproduction/.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures




Similar articles
-
CaLMPhosKAN: prediction of general phosphorylation sites in proteins via fusion of codon aware embeddings with amino acid aware embeddings and wavelet-based Kolmogorov-Arnold network.Bioinformatics. 2025 Mar 29;41(4):btaf124. doi: 10.1093/bioinformatics/btaf124. Bioinformatics. 2025. PMID: 40116777 Free PMC article.
-
GOLabeler: improving sequence-based large-scale protein function prediction by learning to rank.Bioinformatics. 2018 Jul 15;34(14):2465-2473. doi: 10.1093/bioinformatics/bty130. Bioinformatics. 2018. PMID: 29522145
-
learnMSA2: deep protein multiple alignments with large language and hidden Markov models.Bioinformatics. 2024 Sep 1;40(Suppl 2):ii79-ii86. doi: 10.1093/bioinformatics/btae381. Bioinformatics. 2024. PMID: 39230690 Free PMC article.
-
Recent Advances in Computational Prediction of Secondary and Supersecondary Structures from Protein Sequences.Methods Mol Biol. 2025;2870:1-19. doi: 10.1007/978-1-0716-4213-9_1. Methods Mol Biol. 2025. PMID: 39543027 Review.
-
Modern machine learning methods for protein property prediction.Curr Opin Struct Biol. 2025 Feb;90:102990. doi: 10.1016/j.sbi.2025.102990. Epub 2025 Jan 28. Curr Opin Struct Biol. 2025. PMID: 39881454 Review.
Cited by
-
Antibody design using LSTM based deep generative model from phage display library for affinity maturation.Sci Rep. 2021 Mar 12;11(1):5852. doi: 10.1038/s41598-021-85274-7. Sci Rep. 2021. PMID: 33712669 Free PMC article.
-
PEZy-miner: An artificial intelligence driven approach for the discovery of plastic-degrading enzyme candidates.Metab Eng Commun. 2024 Sep 5;19:e00248. doi: 10.1016/j.mec.2024.e00248. eCollection 2024 Dec. Metab Eng Commun. 2024. PMID: 39310048 Free PMC article.
-
Neural network extrapolation to distant regions of the protein fitness landscape.bioRxiv [Preprint]. 2023 Nov 9:2023.11.08.566287. doi: 10.1101/2023.11.08.566287. bioRxiv. 2023. Update in: Nat Commun. 2024 Jul 30;15(1):6405. doi: 10.1038/s41467-024-50712-3. PMID: 37987009 Free PMC article. Updated. Preprint.
-
Identifying Pupylation Proteins and Sites by Incorporating Multiple Methods.Front Endocrinol (Lausanne). 2022 Apr 26;13:849549. doi: 10.3389/fendo.2022.849549. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35557849 Free PMC article.
-
DTI-BERT: Identifying Drug-Target Interactions in Cellular Networking Based on BERT and Deep Learning Method.Front Genet. 2022 Jun 8;13:859188. doi: 10.3389/fgene.2022.859188. eCollection 2022. Front Genet. 2022. PMID: 35754843 Free PMC article.
References
-
- Abbasi W.A., Minhas F.U.A.A. (2016) Issues in performance evaluation for host-pathogen protein interaction prediction. J. Bioinform. Comput. Biol., 14, 1650011.. - PubMed
-
- Alipanahi B. et al. (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol., 33, 831–838. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
