

Elevated polygenic burden for autism is associated with differential DNA methylation at birth
- PMID: 29587883
- PMCID: PMC5872584
- DOI: 10.1186/s13073-018-0527-4
Elevated polygenic burden for autism is associated with differential DNA methylation at birth
Abstract
Background: Autism spectrum disorder (ASD) is a severe neurodevelopmental disorder characterized by deficits in social communication and restricted, repetitive behaviors, interests, or activities. The etiology of ASD involves both inherited and environmental risk factors, with epigenetic processes hypothesized as one mechanism by which both genetic and non-genetic variation influence gene regulation and pathogenesis. The aim of this study was to identify DNA methylation biomarkers of ASD detectable at birth.
Methods: We quantified neonatal methylomic variation in 1263 infants-of whom ~ 50% went on to subsequently develop ASD-using DNA isolated from archived blood spots taken shortly after birth. We used matched genotype data from the same individuals to examine the molecular consequences of ASD-associated genetic risk variants, identifying methylomic variation associated with elevated polygenic burden for ASD. In addition, we performed DNA methylation quantitative trait loci (mQTL) mapping to prioritize target genes from ASD GWAS findings.
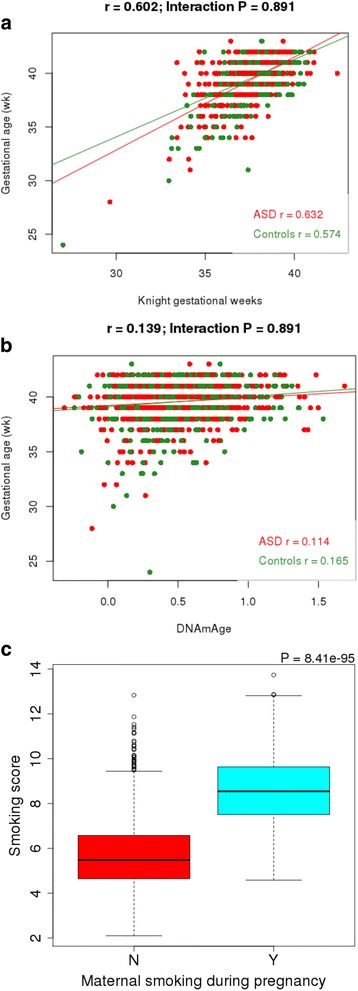
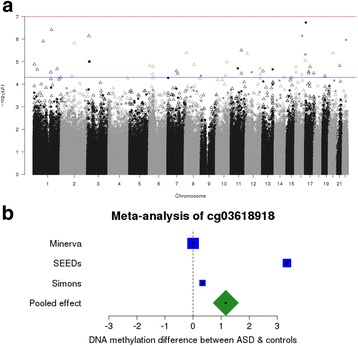
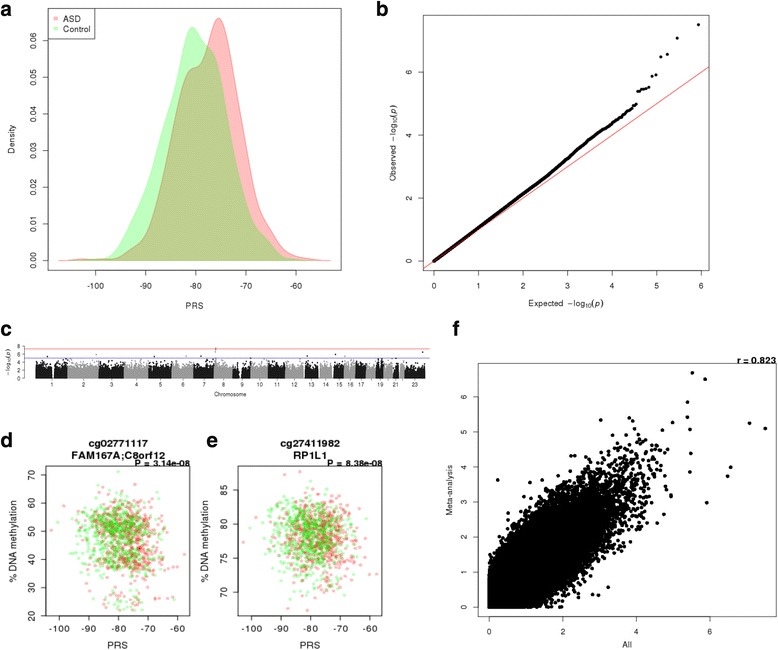
Results: We identified robust epigenetic signatures of gestational age and prenatal tobacco exposure, confirming the utility of DNA methylation data generated from neonatal blood spots. Although we did not identify specific loci showing robust differences in neonatal DNA methylation associated with later ASD, there was a significant association between increased polygenic burden for autism and methylomic variation at specific loci. Each unit of elevated ASD polygenic risk score was associated with a mean increase in DNA methylation of - 0.14% at two CpG sites located proximal to a robust GWAS signal for ASD on chromosome 8.
Conclusions: This study is the largest analysis of DNA methylation in ASD undertaken and the first to integrate genetic and epigenetic variation at birth. We demonstrate the utility of using a polygenic risk score to identify molecular variation associated with disease, and of using mQTL to refine the functional and regulatory variation associated with ASD risk variants.
Keywords: Autism; Birth; DNA methylation; DNA methylation quantitative trait loci (mQTL); Epigenome-wide association study (EWAS); Genetics; Genome-wide association study (GWAS); Neonatal; Polygenic risk score; Prenatal smoking.
Conflict of interest statement
Ethics approval and consent to participate
The MINERvA study has been approved by the Regional Scientific Ethics Committee in Denmark, the Danish Data Protection Agency and the NBS-Biobank Steering Committee. iPSYCH is a register-based cohort study solely using data from national health registries. The study was approved by the Scientific Ethics Committees of the Central Denmark Region (
Consent for publication
Not applicable.
Competing interests
TW has acted as advisor and lecturer to H. Lundbeck A/S. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Washington, DC: The American Psychiatric Association; 2000.
-
- Investigators ADDMNSYP, CfDCa P. Prevalence of autism spectrum disorders--Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ. 2012;61:1–19. - PubMed
-
- Christensen DL, Baio J, Van Naarden BK, Bilder D, Charles J, Constantino JN, Daniels J, Durkin MS, Fitzgerald RT, Kurzius-Spencer M, et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years--Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill Summ. 2016;65:1–23. doi: 10.15585/mmwr.ss6503a1. - DOI - PMC - PubMed
-
- Robinson EB, St Pourcain B, Anttila V, Kosmicki JA, Bulik-Sullivan B, Grove J, Maller J, Samocha KE, Sanders SJ, Ripke S, et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat Genet. 2016;48:552–555. doi: 10.1038/ng.3529. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
