

A single-cell hematopoietic landscape resolves 8 lineage trajectories and defects in Kit mutant mice
- PMID: 29588278
- PMCID: PMC5969381
- DOI: 10.1182/blood-2017-12-821413
A single-cell hematopoietic landscape resolves 8 lineage trajectories and defects in Kit mutant mice
Abstract
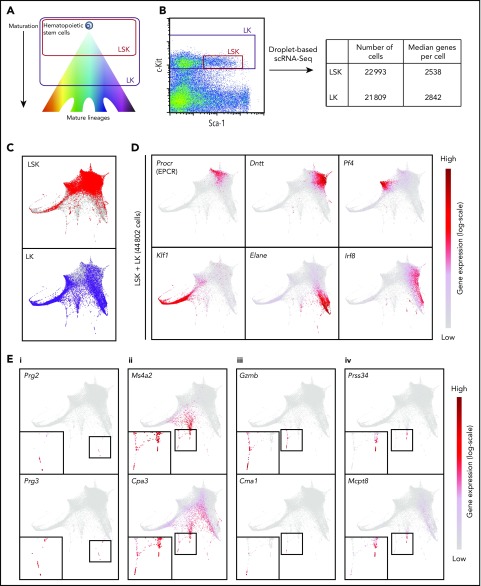
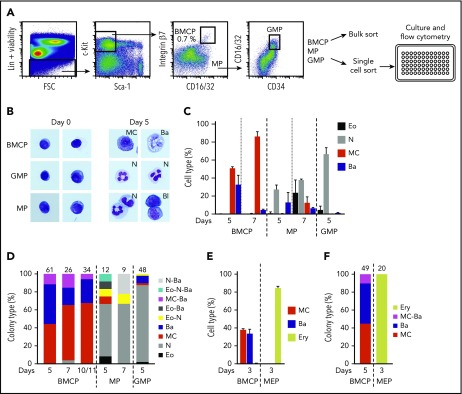
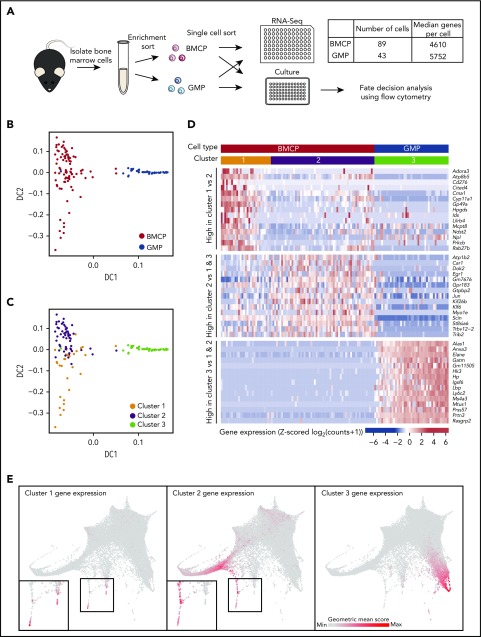
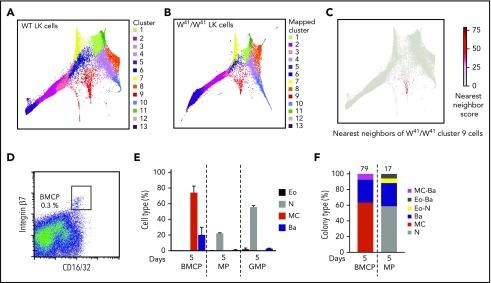
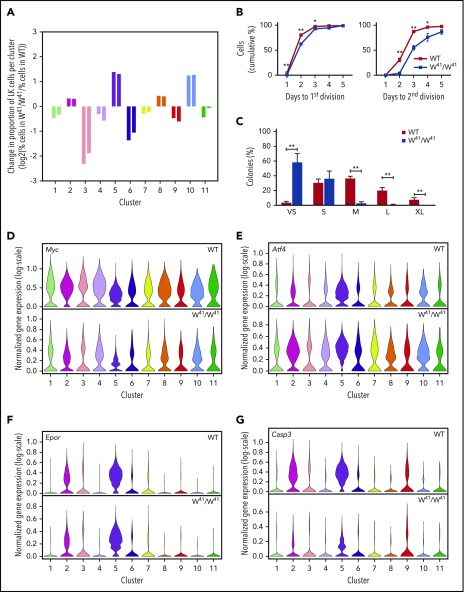
Hematopoietic stem and progenitor cells (HSPCs) maintain the adult blood system, and their dysregulation causes a multitude of diseases. However, the differentiation journeys toward specific hematopoietic lineages remain ill defined, and system-wide disease interpretation remains challenging. Here, we have profiled 44 802 mouse bone marrow HSPCs using single-cell RNA sequencing to provide a comprehensive transcriptional landscape with entry points to 8 different blood lineages (lymphoid, megakaryocyte, erythroid, neutrophil, monocyte, eosinophil, mast cell, and basophil progenitors). We identified a common basophil/mast cell bone marrow progenitor and characterized its molecular profile at the single-cell level. Transcriptional profiling of 13 815 HSPCs from the c-Kit mutant (W41/W41) mouse model revealed the absence of a distinct mast cell lineage entry point, together with global shifts in cell type abundance. Proliferative defects were accompanied by reduced Myc expression. Potential compensatory processes included upregulation of the integrated stress response pathway and downregulation of proapoptotic gene expression in erythroid progenitors, thus providing a template of how large-scale single-cell transcriptomic studies can bridge between molecular phenotypes and quantitative population changes.
© 2018 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
References
-
- Ema H, Morita Y, Suda T. Heterogeneity and hierarchy of hematopoietic stem cells. Exp Hematol. 2014;42(2):74-82.e2. - PubMed
-
- Adolfsson J, Månsson R, Buza-Vidas N, et al. . Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121(2):295-306. - PubMed
-
- Paul F, Arkin Y, Giladi A, et al. . Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell. 2015;163(7):1663-1677. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
