

Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis
- PMID: 29588315
- PMCID: PMC6160368
- DOI: 10.1161/CIRCULATIONAHA.117.032636
Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis
Abstract
Background: The CANTOS trial (Canakinumab Antiinflammatory Thrombosis Outcome Study) showed that antagonism of interleukin (IL)-1β reduces coronary heart disease in patients with a previous myocardial infarction and evidence of systemic inflammation, indicating that pathways required for IL-1β secretion increase cardiovascular risk. IL-1β and IL-18 are produced via the NLRP3 inflammasome in myeloid cells in response to cholesterol accumulation, but mechanisms linking NLRP3 inflammasome activation to atherogenesis are unclear. The cholesterol transporters ATP binding cassette A1 and G1 (ABCA1/G1) mediate cholesterol efflux to high-density lipoprotein, and Abca1/g1 deficiency in myeloid cells leads to cholesterol accumulation.
Methods: To interrogate mechanisms connecting inflammasome activation with atherogenesis, we used mice with myeloid Abca1/g1 deficiency and concomitant deficiency of the inflammasome components Nlrp3 or Caspase-1/11. Bone marrow from these mice was transplanted into Ldlr-/- recipients, which were fed a Western-type diet.
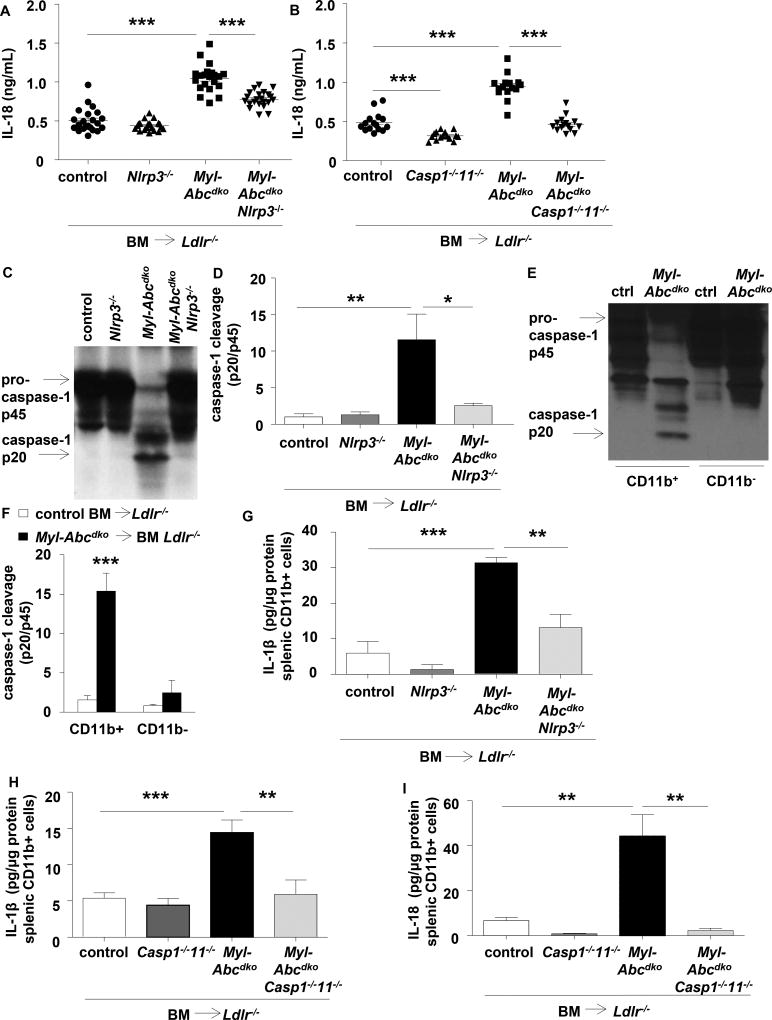
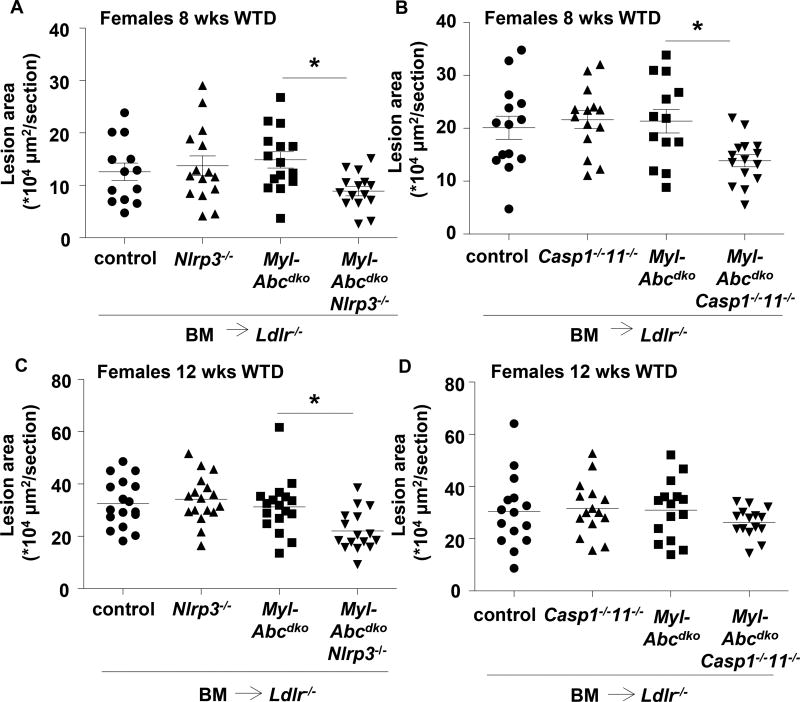
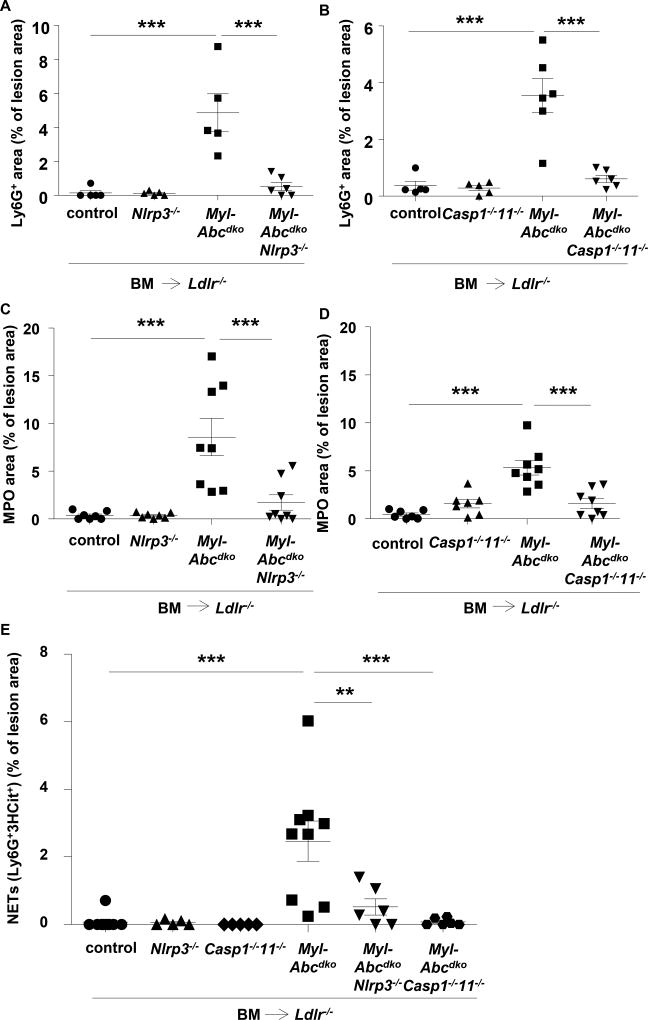
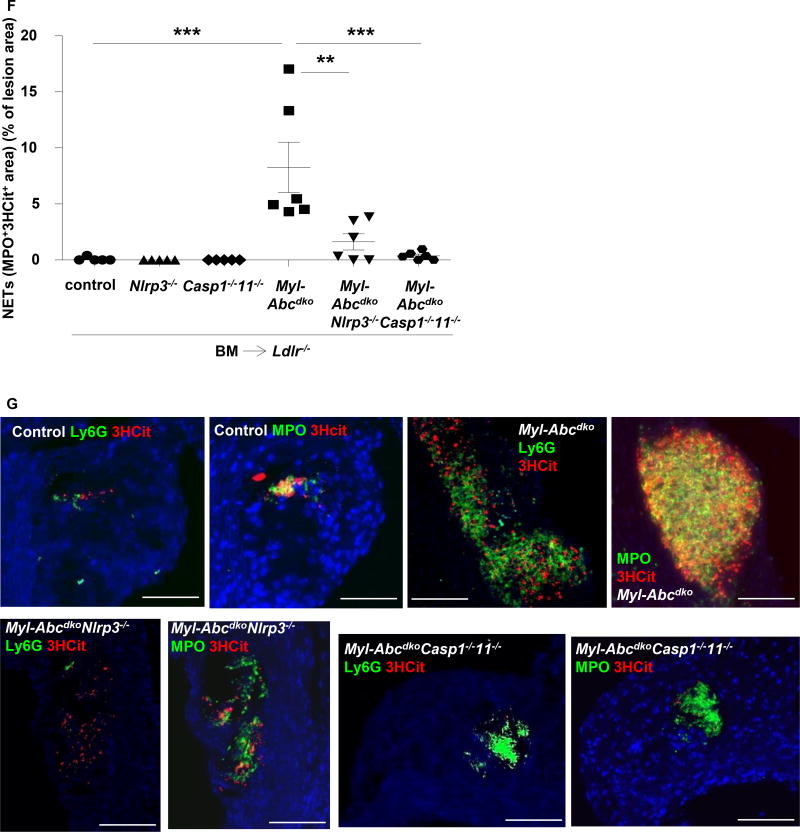
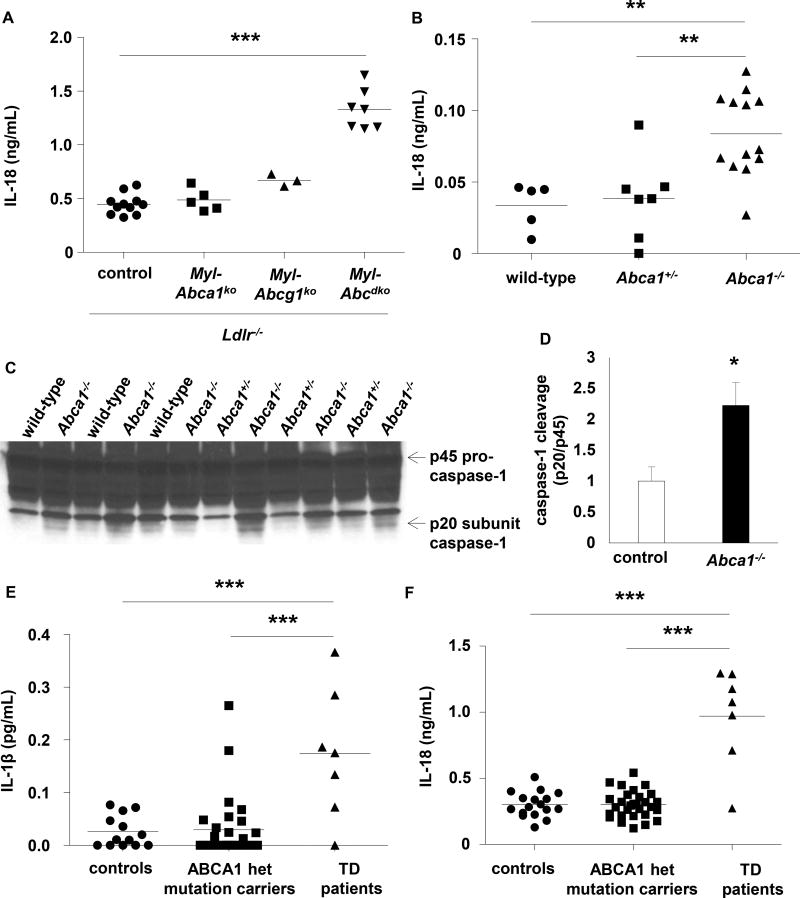
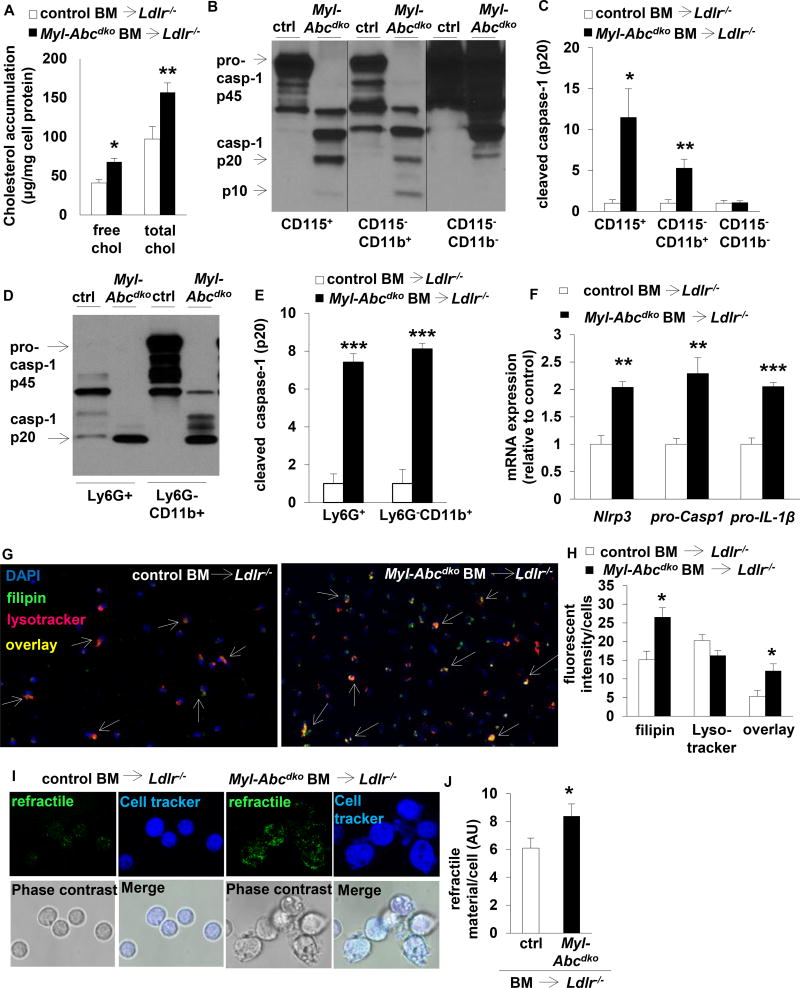
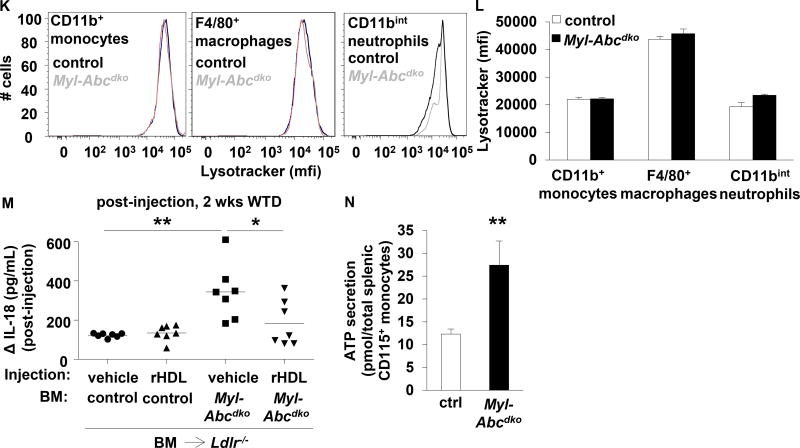
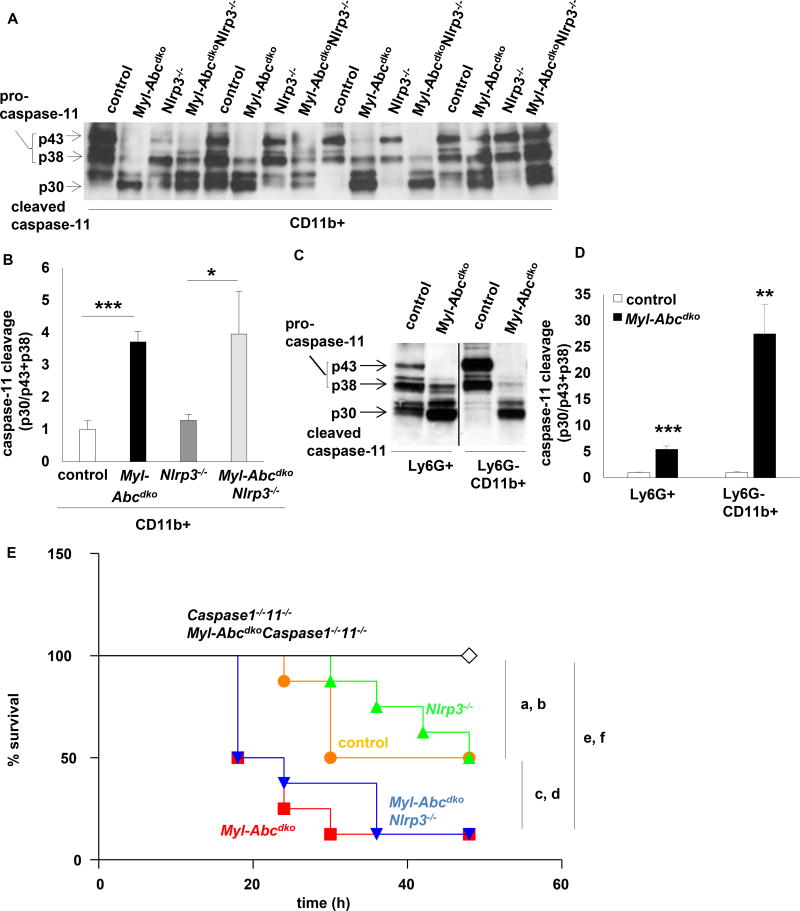
Results: Myeloid Abca1/g1 deficiency increased plasma IL-18 levels in Ldlr-/- mice and induced IL-1β and IL-18 secretion in splenocytes, which was reversed by Nlrp3 or Caspase-1/11 deficiency, indicating activation of the NLRP3 inflammasome. Nlrp3 or Caspase-1/11 deficiency decreased atherosclerotic lesion size in myeloid Abca1/g1-deficient Ldlr-/- mice. Myeloid Abca1/g1 deficiency enhanced caspase-1 cleavage not only in splenic monocytes and macrophages, but also in neutrophils, and dramatically enhanced neutrophil accumulation and neutrophil extracellular trap formation in atherosclerotic plaques, with reversal by Nlrp3 or Caspase-1/11 deficiency, suggesting that inflammasome activation promotes neutrophil recruitment and neutrophil extracellular trap formation in atherosclerotic plaques. These effects appeared to be indirectly mediated by systemic inflammation leading to activation and accumulation of neutrophils in plaques. Myeloid Abca1/g1 deficiency also activated the noncanonical inflammasome, causing increased susceptibility to lipopolysaccharide-induced mortality. Patients with Tangier disease, who carry loss-of-function mutations in ABCA1 and have increased myeloid cholesterol content, showed a marked increase in plasma IL-1β and IL-18 levels.
Conclusions: Cholesterol accumulation in myeloid cells activates the NLRP3 inflammasome, which enhances neutrophil accumulation and neutrophil extracellular trap formation in atherosclerotic plaques. Patients with Tangier disease, who have increased myeloid cholesterol content, showed markers of inflammasome activation, suggesting human relevance.
Keywords: ATP-binding cassette transporters; atherosclerosis; cholesterol, HDL; extracellular traps; inflammasomes.
Conflict of interest statement
A.R.T. reports being a consultant to Amgen, CSL, Staten Biotech, and Dalcor. The other authors report no conflicts of interest.
Figures
Comment in
-
Cholesterol-dependent inflammasome activation accelerates atherosclerosis.Nat Rev Cardiol. 2018 Jun;15(6):318-319. doi: 10.1038/s41569-018-0012-1. Nat Rev Cardiol. 2018. PMID: 29674715 No abstract available.
-
High-density lipoprotein lifts the "dark web" cast by neutrophils.Ann Transl Med. 2018 Nov;6(Suppl 1):S24. doi: 10.21037/atm.2018.09.28. Ann Transl Med. 2018. PMID: 30613599 Free PMC article. No abstract available.
References
-
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Group CT. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. - PubMed
-
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. - PMC - PubMed
-
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andres V, Hirschi KK, Martin KA, Walsh K. Clonal hematopoiesis associated with tet2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. - PMC - PubMed
-
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. - PMC - PubMed
-
- Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, Bayard F, Hansson GK. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein e-knockout mice. Cardiovasc Res. 2003;59:234–240. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
